Introduction
Diabetes mellitus is seen as one of the main threats to human health. The past two decades have seen an explosive increase in the number of people diagnosed worldwide, affecting, nowadays, about 250 millions of people. In addition, it has been estimated that in 2025 about 380 millions of people will be suffering from this disease (Hegart et al. 2009).
Diabetes mellitus can be caused by destruction of pancreatic β-cells islets due to an autoimmune attack on these cells, characterizing diabetes mellitus type 1 (DM1) (Eizirik et al. 2009). However, the most common form of diabetes mellitus, known as diabetes mellitus type 2 (DM2), results from a sequence of events. In a person with pancreatic β-cells responding normally, an increase in glycaemia is accompanied by a transitory increase in insulinemia, with glycaemia returning to baseline values. In people prone to developing DM2, the failure of compensation in pancreatic β-cells (associated or not with peripheral insulin resistance and/or genetic predisposition) maintain glycaemia in a chronically elevated state. Consequently, in the course of time, the association of glucotoxicity and lipotoxicity promote a failure/destruction of pancreatic β-cells, reducing its mass, and difficulting even more the process of glycaemic control (Eizirik and Cnop, 2010; Guillausseau et al. 2008; Kahn, 2003; Poitout and Robertson, 2002).
Due to the role of pancreatic β-cells in diabetes, therapies that support their functionality are necessary. Dehydroepiandrosterone (DHEA) is a steroid hormone synthesized and secreted by adrenal gland cortex, and shows an oscillatory rhythm (high and low levels) in the circulation. Its peak occurs in the third decade of life, and it decays from 40 years old (Rainey and Nakamura, 2008; Rainey et al. 2002). In accordance with it, it is also observed an increasing incidence of several chronic-degenerative diseases in the population, including DM2. In this sense, many researchers sought to study the relationship between DHEA and insulin resistance (Weiss et al. 2011), vascular functionality (Camporez et al. 2011), adipose tissue (Fujioka et al. 2012), neuroprotection (Aly et al. 2011), among others.
In addition, previous data from our laboratory demonstrated an increase in insulin secretory capacity in pancreatic islets of rats with 12-14 months-old that received only one dose of DHEA (Medina et al. 2006). Therefore, with this research, we intended to identify the effects of incubation with DHEA on insulin secretory and cytoprotective capacity in INS-1E beta cells.
Materials and Methods
INS-1E Cell Culture
The INS-1E cell line was cultured as previously described (Merglen et al. 2004). INS-1E cells were cultured in a humidified atmosphere containing 5% CO2 in complete medium composed of RPMI 1640 supplemented with heat-inactivated fetal calf serum, 1mM sodium pyruvate, 50 µM 2-mercaptoethanol, 2 mM glutamine, 10 mM HEPES, 100 U/ml penicillin, and 100 µg/ml streptomycin. In DHEA treated cells, the culture medium was replaced by a medium with DHEA in different concentrations (10 µM, 100 nM, and 1 nM). Control group was incubated with vehicle (DMSO). Two different periods of incubations were used (1 and 7 days) (Santos et al. 2011).
Insulin Secretion
INS-1E cells were starved in RPMI medium without glucose for 2 h. After this period, cells were preincubated for 30 min at 2.8 mM glucose in Krebs-Henseleit (KH) buffer [in mM: 115 NaCl, 24 NaHCO3, 5 KCl, 1 MgCl2, 1 CaCl2 (pH 7.4)] supplemented with 0.1% BSA and equilibrated in a mixture of O2 (95%) and CO2 (5%) at 37ºC. After that, the medium was changed, and INS-1E cells were incubated for an additional 60 min in the absence or presence of 2.8 and 16.7 mM glucose. At the end of the experiment, the supernatant was collected, and the total insulin content was obtained by disrupting cells in a mixture of ethanol, water, and HCl (52:17:1, vol/vol). Insulin secretion and insulin content were measured by RIA (Santos et al. 2011). DNA content was also determined using NanoDrop. The results of insulin secretion were calculated as secreted insulin per DNA content.
Determination of Superoxide Content
Cells were preincubated at 37ºC for 30 min in KH buffer at 2.8 mM glucose. After that, the cells were incubated for 40 min in KH buffer at 2.8 or 16.7 mM glucose. After incubation, the cells were loaded with 25 μM hydroethidine (HE) and incubated for 20 min at room temperature. INS-1E superoxide content was evaluated by flow cytometry (Guava; Millipore, Billerica, MA) (Santos et al. 2011).
Western Blot Analysis
Total protein content of INS-1E cells was extracted in protein extraction buffer (100 mM Tris, 1% sodium dodecyl sulfate, 10 mM EDTA, 100 mM Na2P2O7, 100 mM NaF, 10 mM Na2VO4). The extracts were centrifuged at 12.900 × g for 20 min at 4ºC. The supernatant was used to protein quantification that was performed by the Bradford method. Proteins were treated with Laemilli buffer, subjected to SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked for 2 hours, and incubated with primary antibodies IRS-2, ERK ½, pERK ½, pJNK, PDX-1 (Santa Cruz Biotechnology), and cleaved caspase-3 (Upstate Biotechnology).
Statistical Analysis
Results are presented as mean ± SEM, and the statistical analyses were carried out using one-way ANOVA (comparisons between three or more groups) with Tukey’s test multiple comparisons. Statistical significance was set to P < 0.05.
Results
Glucose-Stimulated Insulin Secretion
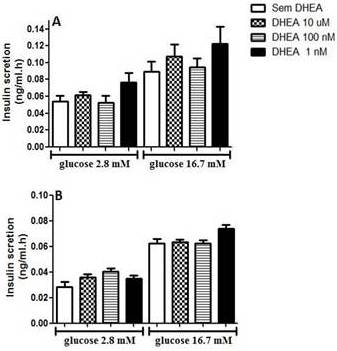
In figure 1, we present data from glucose-stimulated insulin secretion in insulin-secreting INS-1E beta cells incubated with DHEA for 24 hours and 7 days. As one can observe, DHEA incubation for 24 hours (Figure 1A) or 7 days (Figure 1 B) did not reflect in insulin secretion capacity of INS-1E cells.
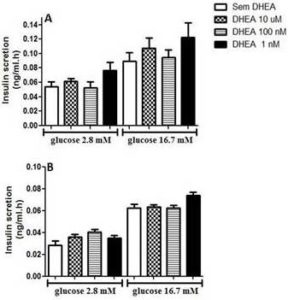
Figure 1. Glucose-Stimulated Insulin Secretion of INS-1E Cells Incubated with Different Concentration of Dehydroepiandrosterone for (A) 24 Hours, and (B) 7 Days. Values were Corrected by DNA Content. (N=5-8 Different Experiments).
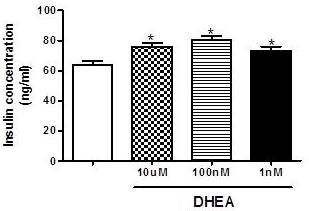
In contrast, as observed in figure 2, total insulin content after insulin secretion assay is higher in cells incubated with different concentrations of DHEA (10 µM, 100 nM and 1 nM), and this could be an indicator of increased insulin production.
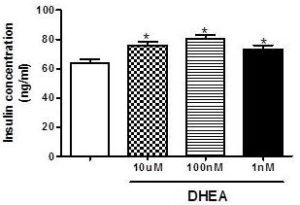
Figure 2. Total Insulin Content after Glucose-Stimulated Insulin Secretion Assay in INS-1E after 7 Days of Incubation with Different Concentrations of DHEA. *P<0.05 in Relation to Control Group. (N=5-8 Different Experiments).
Superoxide Production
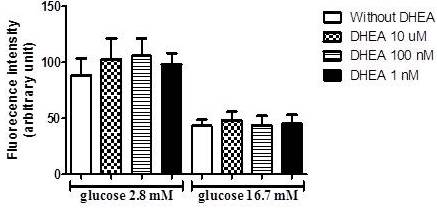
Afterward, due to known antioxidants effects of DHEA and the influence of reactive oxygen species on insulin secretion, we perform an assay to verify superoxide production in INS-1E cells incubated with low (2.8 mM) and high (16.7 mM) glucose concentrations. As observed in figure 3, the incubation with DHEA for 7 days did not influence superoxide production.
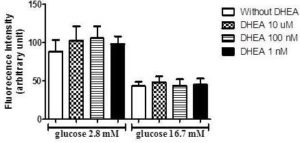
Figure 3. DHEA Does Not Change Superoxide Content in INS-1E Cells after 1 Hour of Incubation with Low (2.8 Mm), and High (16.7 Mm) Glucose. (N=5-8 Different Experiments).
Protein Concentration
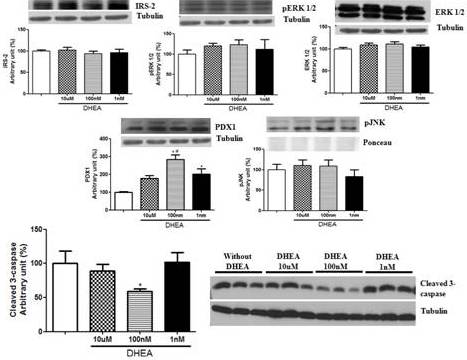
In addition to secretion assays, we extracted the proteins and performed western blotting to analyze some protein concentration that could help us to explain our results. In this sense, proteins involved in cell proliferation pathways (IRS-2, pERK1/2 and ERK1/2), apoptosis (pJNK and cleaved caspase-3) and insulin synthesis/development and maturation of pancreatic β-cells (PDX1) were evaluated (Figure 4). The treatment with DHEA for 7 days did not change the levels of any protein involved in cell proliferation. On the other hand, it reduced cleaved caspase-3 expression (at 100 nM concentration of DHEA) and increased protein concentration of PDX1 (at 100 nM and 1 nM, and also a strong tendency at 10 µM concentrations of DHEA).
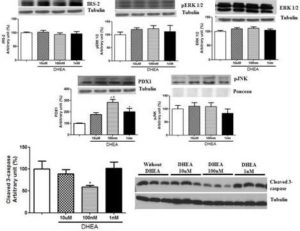
Figure 4. Protein Concentration of IRS-2, Perk 1/2, ERK 1/2, PDX1, Pjnk, and Cleaved 3-Caspase of INS-1E Cells Obtained by Western Blotting. Cells were Incubated with Different Concentrations of DHEA for 7 Days. *P<0.05 in Relation to Control Group. #P<0.05 in Relation to 10 µm and 1 Nm Groups.
Discussion
The present work sought to identify the effects of DHEA treatment on insulin secretory and cytoprotective capacity in INS-1E cells. As observed, DHEA incubations did not resulted in an increased insulin secretory capacity but increases insulin content, PDX-1 concentration and reduced cleaved 3-caspase.
Although the relation between the reduction of DHEA circulating levels and chronic-degenerative diseases like obesity, insulin resistance and diabetes mellitus type 2, few studies aimed to study the effects of DHEA on insulin secretory capacity. Ladriele et al. (1997) identified that the treatment of Goto-Kakizaki rats with DHEA improved the relation insulinemia/glycaemia indicating a greater insulin sensibility, associated to a greater sensibility of pancreatic islets to glucose. Additionally, Dillon et al. (2000) demonstrated that the treatment with sulfated form of DHEA (DHEA-S) for 3 and 7 days increased insulin secretory capacity in pancreatic islets, INS-1E and HIT-T15 cells. These authors indicated that these increases did not occur with 24-hour treatment, so the action was not direct, but it was mediated by gene alteration (they observed an increased gene expression of compounds involved in mitochondrial and peroxisomal lipid oxidation). Posteriorly, data from our laboratory (Medina et al. 2006) identified an increased insulin secretory capacity in pancreatic isles of rats treated with DHEA. Thus, our data in INS-1E cells do not corroborate those from literature demonstrating a new vision in this topic.
When analyzing total insulin content, we observe that DHEA treatment, independently of the concentration, increased the amount of total insulin in INS-1E cells (~10%). Probably, DHEA improves the machinery involved in this process. The higher insulin content could be reflected of an elevated number of cells. In this sense, analyzing protein concentration we sought to identify the pathways in which this changes occurred. First of all, we analyzed PDX1. PDX1 is the greater regulator of glucose-stimulated gene transcription, it is essential to the embrionary development of pancreas; it plays a central role in maturation and functioning of pancreatic β-cell in adult life, and is related with pancreatic β-cell apoptosis (Fujimoto and Polonsky, 2009; Andrali et al. 2008; Kaneto et al. 2007). As observed, DHEA treatment elevated the concentration of PDX1 (100nM and 1nM, p<0.05; 10µM – tendency of statistical significance), what could be one of the causes for the increase in insulin content and/or pancreatic β-cell.
In addition, we investigated protein concentration of IRS-2, due to its participation in the stimulation of pancreatic islet development (Kubota et al. 2000; Whiters et al. 1998). Previous data from our laboratory observed a reduction in protein concentration of IRS-2 in pancreatic islets of rats treated with DHEA (Medina et al. 2006). However, in the incubations in vitro with DHEA in INS-1E cells, we did not observe any change in protein concentration of IRS-2. Other protein analyzed, and that is associated with pancreatic β-cells proliferation and with insulin gene transcription, is ERKs 1 and 2 (Lawrence et al. 2008). Once again, DHEA treatment did not act on ERK1/2 protein concentration and in its phosphorylated form pERK1/2.
Another mechanism that could be related with the increase in pancreatic β-cells concentration is the inhibition of apoptosis pathways, autophagia and necrosis (Scarlatti et al. 2009). Apoptosis is the major cause of cell death in pancreatic β-cells (Chang-Chen et al. 2008; Butler et al. 2007). In this sense, we sought to identify protein concentration of cleaved caspase-3, a final effector of apoptosis pathway (Tomita, 2010). We observed that with DHEA treatment, at the concentration of 100 nM, reduced protein concentration of cleaved caspase-3, indicating a cytoprotective effect of DHEA. This activity of DHEA is interesting, since the reduction of the amount of pancreatic β-cells is one key characteristic in the development of DM2 (Cnop et al. 2011). Nevertheless, due to the absence of a stimulus triggering cell death, we should be cautious about this conclusion, since it can result in tumor growth.
Finally, due to some DHEA effects being mediated by changes in cell oxidative balance (Huerta-Garcia et al. 2012; Camporez et al. 2011) and, the influence of oxygen reactive species in insulin secretory capacity (Rebelato et al. 2011), we verified if DHEA treatment would result in changes of superoxide production in INS-1E cells, but we did not found any influence.
In conclusion, the incubations of pancreatic β-cells (INS-1E line) with DHEA increased insulin content, without effects in insulin secretion. This effect can be observed in this research with 7 days of incubation. Partly, the mechanism responsible for this greater insulin content should be the protein concentration of PDX1. Another important result involves the cytoprotective effect of DHEA at the concentration of 100 nM. These results indicate that different concentrations could reflect in different effects. Thus, other studies are necessary to investigate these potential effects in higher PDX-1/insulin production and reduced cleaved-caspase 3/cell apoptosis of DHEA.
References
Aly, H. F., Metwally, F. M. & Ahmed, H. H. (2011). “Neuroprotective Effects of Dehydroepiandrosterone (DHEA) in Rat Model of Alzheimer’s Disease,” Acta Biochimica Polonica, 58: 513-20.
Publisher – Google Scholar
Andrali, S. S., Sampley, M. L., Vanderford, N. L. & Ozcan, S. (2008). “Glucose Regulation of Insulin Gene Expression in Pancreatic Beta-Cell,” Biochemical Journal, 415: 1-10.
Publisher – Google Scholar
Camporez, J. P. G., Akamine, E. H., Davel, A. P., Franci, C. R., Rossoni, L. V. & Carvalho, C. R. O. (2011). “Dehydroepiandrosterone Protects Against Oxidative Stress-Induced Endothelial Dysfunction in Ovariectomized Rats,”The Journal of Physiology, 589: 2585-96.
Publisher – Google Scholar
Chang-Chen, K. J., Mullur, R. & Bernal-Mizrachi, E. (2008). “Beta-Cell Failure as Complication of Diabetes,” Reviews in Endocrine and Metabolic Disorders, 9: 329-43.
Publisher – Google Scholar
Cnop, M., Igoillo-Esteve, M., Hughes, S. J., Walker, J. N., Cnop, I. & Clark, A. (2011). “Longevity of Human Islet Alfa- and Beta-Cells,” Diabetes, Obesity and Metabolism, Suppl 1: 39-46.
Publisher – Google Scholar
Dillon, J. S., Yaney, G. C., Zhou, Y., Voilley, N., Bowen, S., Chipkin, S., Bliss, C. R., Schultz, V., Schuit, F. C., Prentki, M.,Waxman, D. J. & Corkey, B. E. (2000). “Dehydroepiandrosterone Sulfate and Beta Cell Function: Enhanced Glucose-Induced Insulin Secretion and Altered Gene Expression in Rodent Pancreatic Beta-Cells,” Diabetes, 49: 2012-20.
Publisher – Google Scholar
Eizirik, D. L. & Cnop, M. (2010). “ER Stress in Pancreatic Beta Cells: The Thin Red Line between Adaptation and Failure,” Science Signaling, 3: 1-4.
Publisher – Google Scholar
Eizirik, D. L., Colli, M. L. & Ortis, F. (2009). “The Role of Inflammation in Insulitis and Beta-Cell Loss in Type 1 Diabetes,”Nature Reviews Endocrinology, 5: 219-26.
Publisher – Google Scholar
Fujimoto, K. & Polonsky, K. S.(2009). “Pdx 1 and Other Factors That Regulate Pancreatic Beta-Cell Survival,” Diabetes, Obesity and Metabolism, Suppl 4: 30-7.
Publisher – Google Scholar
Fujioka, K., Kajita, K., Wu, Z., Hanamoto, T., Ikeda, T., Mori, I., Okada, H., Yamauchi, M., Uno, Y., Morita, H., Nagano, I., Takahashi, Y. & Ishizuka, T. (2012). “Dehydroepiandrosterone Reduces Preadipocyte Proliferation Via Androgen Receptor,” American Journal of Physiology – Endocrinology and Metabolism, 302: 694-704.
Publisher – Google Scholar
Guillausseau, P. J., Meas, T., Virraly, M., Laloi-Michelin, M., Médeau, V. & Kevorkian, J. P. (2008). “Abnormalities in Insulin Secretion in Type 2 Diabetes Mellitus,” Diabetes & Metabolism, 34: S43-8.
Publisher – Google Scholar
Hegart, B. D., Turner, N., Cooney, G. J. & Kraegen, E. W. (2009). “Insulin Resistance and Fuel Homeostasis: The Role of AMP-Activated Protein Kinase,” Acta Physiologica, 196: 129-45.
Publisher – Google Scholar
Kahn, S. E. (2003). “The Relative Contributions of Insulin Resistance and Beta-Cell Dysfunction to the Pathophysiology of Type 2 Diabetes,” Diabetologia, 46: 3-19.
Publisher – Google Scholar
Kaneto, H., Miyatsuka, T., Fujitani, Y., Noguchi, H., Song, K., Yoon, K. & Matsuoka, T. A. (2007). “Role of PDX1 and Mafa as Potential Therapeutic Target for Diabetes,” Diabetes Research and Clinical Practice, 77S: S127-37.
Publisher – Google Scholar
Kubota, N., Tobe, K., Terauchi, Y., Eto, K., Yamauchi, T., Suzuki, R., Tsubamoto, Y., Komeda, K., Nakano, R., Miki, H., Satoh, S., Sekihara, H., Sciacchitano, S., Lesniak, M., Aizawa, S., Nagai, R., Kimura, S., Akanuma, Y., Taylor, S. I. & Kadowaki, T. (2000). “Disruption of Insulin Receptor Substrate 2 Causes Type 2 Diabetes Because of Liver Insulin Resistance and Lack of Compensatory Beta-Cell Hyperplasia,” Diabetes, 49: 1880-9.
Publisher – Google Scholar
Ladriere, L., Laghmich, A., Malaisse-Lagae, F. & Malaisse, W. J. (1997). “Effect of Dehydroepiandrosterone in Hereditarily Diabetic Rats,” Cell Biochemistry and Function, 15: 287-91.
Publisher – Google Scholar
Lawrence, M., Shao, C., Duan, L., Mcglynn, K. & Cobb, M. H. (2008). “The Protein Kinas ERK1/2 and Their Roles in Pancreatic Beta Cells,” Acta Physiologica (Oxf), 192: 11-7.
Publisher – Google Scholar
Medina, M. C., Souza, L. C., Caperuto, L. C., Anhe, G. F., Amanso, A. M., Teixeira, V. P. A., Bordin S., Carpinelli, A. R., Brito, L. R., Barbieri, R. L., Borella, M. I. & Carvalho, C. R. (2006). “Dehydroepiandrosterone Increases Beta-Cell Mass and Improves the Glucose-Induced Insulin Secretion by Pancreatic Islets from Aged Rats,” FEBS Letters, 580: 285-90.
Publisher – Google Scholar
Merglen, A., Theander, S., Rubi, B., Chaffard, G., Wollheim, C. B. & Maechler, P. (2004). “Glucose Sensitivity and Metabolism-Secretion Coupling Studied During Two-Year Continuous Culture in INS-1E Insulinoma Cells,”Endocrinology, 145: 667-78.
Publisher – Google Scholar
Poitout, V. & Robertson, R. P. (2002). “Secondary Beta-Cell Failure in Type 2 Diabetes: A Convergence of Glucotoxicity and Lipotoxicity,” Endocrinology, 143: 339-42.
Publisher – Google Scholar
Rainey, W. E., Carr, B. R., Sasano, H., Suzuki, T. & Mason, I. (2002). “Dissecting Human Adrenal Androgen Production,”Trends in Endocrinology and Metabolism, 13: 234-39.
Publisher – Google Scholar
Rainey, W. E. & Nakamura, Y. (2002). “Regulation of Adrenal Androgen Biosynthesis,” The Journal of Steroid Biochemistry and Molecular Biology, 108: 281-86.
Publisher – Google Scholar
Rebelato, E., Abdulkader, F., Curi, R. & Carpinelli, A. R. (2011). “Control of the Intracellular Redox State by Glucose Participates in the Insulin Secretion Mechanism,” PLOS One, 6: 1-8.
Publisher – Google Scholar
Santos, L. R. B., Rebelato, E., Graciano, M. F. R., Abdulkader, F., Curi, R. & Carpinelli, A. R. (2011). “Oleic Acids Modulates Metabolic Substrate Channeling during Glucose —Stimulated Insulin Secretion Via NAD (P) H Oxidase,”Endocrinology, 152: 3614-21.
Publisher – Google Scholar
Scarlatti, F., Granata, R., Meijer, A. J. & Codogno, P. (2009). “Does Autophagy Have a Licence to Kill Mammalian Cells?,” Cell Death and Differentiation, 16: 12-20.
Publisher – Google Scholar
Tomita, T. (2010). “Immunocytochemical Localisation of Caspase-3 in Pancreatic Islets from Type 2 Diabetic Subjects,”Pathology, 42: 432-37.
Publisher – Google Scholar
Weiss, E. P., Villareal, D. T., Fontana, L., Han, D. H. & Hollosky, J. O. (2011). “Dehydroepiandrosterone (DHEA) Replacement Decreases Insulin Resistance and Lowers Inflammatory Cytokines in Aging Humans,” Aging, 3: 533-42.
Publisher – Google Scholar
Whiters, D. J., Gutierrez, J. S., Towery, H., Burks, D. J., Ren, J., Previs, S., Zhang, Y., Bernal, D., Pons, S., Shulman, G. I., Bonner-Weir, S. & White, M. F. (1998). “Disruption of IRS-2 Causes Type 2 Diabetes in Mice,” Nature, 391: 900-4.
Publisher – Google Scholar