Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) (Namaka et al., 2011b). There are essentially two phases of disease, an inflammatory phase involving the infiltration of active T-cells and monocytes into the CNS, followed by a second phase of disease involving immune mediated cell death of oligodendrocytes (OGs) (Namaka et al., 2011b). Due to the lack of human tissue samples, MS research has largely relied on various animal models (Lu et al., 2012, Stepanov et al., 2012) to explore the intricate cellular signaling that regulates myelin structure, function and repair. Animal models using myelin basic protein (MBP) as the antigen, characteristically activate lymphocytes that mediate an inflammatory response without demyelination (Genain and Hauser, 1996). One of the characteristic pathological features of MS is the identification of segments of demyelinated axons with subsequent death of OGs (Totoiu and Keirstead, 2005, Emery et al., 1998, McTigue et al., 2001, Mekhail et al., 2012). Oligodendrocyte progenitors (OPs) attempt to proliferate and become mobilized for subsequent recruitment to the sites of injury in order to re-myelinate denuded axons (Keirstead et al., 1998, Gensert and Goldman, 1997, McTigue et al., 2001). However, they are unable to completely repair the damaged myelin. The cellular mechanisms that govern this “insufficiency” are currently unknown. Although several studies have shown that OPs exist in the CNS (Horner et al., 2000, Gogate et al., 1994, Frost et al., 2003), their inability to appropriately migrate, proliferate and differentiate at the site of SC injury results in partial or incomplete myelination (McTigue et al., 2001, Lytle et al., 2009, Ishii et al., 2001). Previous studies have tried to target the effects of growth factors on OPs to optimize this process but these efforts have resulted in only marginal success (Messersmith et al., 2000, Armstrong et al., 2002, Frost et al., 2003). Thus, the mechanism for re-myelination of SC white matter is still unknown. Henceforth, the current research is focusing on examining the time dependent expression changes of TrkB during the early inflammatory phase of EAE in the SC as a possible mechanism of cellular responsiveness to local BDNF release.
Regulation of Myelin Formation via BDNF: The neurotrophin BDNF has been suggested as a critical factor involved in re-myelination and/or the structural repair of myelin (Linker et al., 2009, Lykissas et al., 2007). However, the specific molecular mechanism(s) linking BDNF activity to SC myelin repair are not well defined. Furthermore, little is known about the exact molecular mechanisms by which BDNF/TrkB regulates the myelin-producing cells and the structural protein composition of myelin. Irrespective, part of the beneficial effects of BDNF result from its direct ability to induce OP proliferation (Van’t Veer et al., 2009), migration and differentiation at the site(s) of injury (Nakajima et al., 2010), which are essential steps in myelin formation (Vondran et al., 2010, Cellerino et al., 1997). In addition to regulating OG lineage cells, BDNF also regulates other cell types (including astrocytes and neurons) that are also critical for myelination (Emery, 2011). Furthermore, BDNF is responsible for regulating the expression of the main structural proteins of myelin such as MBP (Djalali et al., 2005), myelin associated glycoprotein (MAG), and proteolipid protein (PLP) in mature OGs (Cellerino et al., 1997, Du et al., 2006, Vondran et al., 2010). In addition, we have also published research that provides key evidence in support of BDNF and myelin repair. Our recent study demonstrated that following EAE induction, BDNF was up-regulated in the DRG and delivered to the SC (Zhu et al., 2012). Specifically, small to medium-sized sensory neurons housed within the DRG were identified as the cellular source of BDNF being delivered to the SC (Zhu et al., 2012). As a result, DRG-derived BDNF is now recognized as an essential requirement for SC myelin repair and subsequent neurological recovery following an immune system mediated attack.
BDNF Molecular Signaling: BDNF is synthesized and secreted as a precursor protein, called proBDNF (34 kDa), which can be proteolytically-cleaved to produce a mature form (13 kDa) (Seidah et al., 1996, Pang et al., 2004, Kolbeck et al., 1994). In general, BDNF signaling is regulated via a receptor complex that encompasses two different receptor classes: the tropomyosin-related kinase (Trk) receptor B (TrkB) (Patapoutian and Reichardt, 2001, Bibel et al., 1999) and the p75NTR receptor, a member of the tumor necrosis factor (TNF) receptor superfamily (Bibel et al., 1999, Lee et al., 2001). Previous studies have shown that SC OGs do not express TrkB, but do express p75NTR (Althaus et al., 2008, Cohen et al., 1996). The mature, 13kDa form of BDNF only signals via the TrkB receptor (Patapoutian and Reichardt, 2001), whereas, proBDNF (34 kDa) interacts with the p75 neurotrophin receptor (NTR) (Binder and Scharfman, 2004) to mediate biological actions distinct from those of TrkB (Bibel et al., 1999, Schecterson and Bothwell, 2010, Shamovsky et al., 1999). Although there are no published studies addressing TrkB expression in SC OGs, there are studies showing that proBDNF promotes myelination via p75NTR-mediated activity independent of TrkB (Cosgaya et al., 2002, Teng et al., 2005, Acosta, 2013). Specifically, inhibition of the p75NTR receptor activity prevented the expression of one of the key structural proteins of myelin called MAG (Cosgaya et al., 2002). Thus, proBDNF is critical for the structural integrity of the intact myelin sheath through TrkB-independent pathways. However, despite the apparent beneficial roles of BDNF/TrkB in myelin repair, its exact mechanism is still unknown (Zhu et al., 2012).
We hypothesized that SC BDNF responsive cells contribute to myelin repair and neurological recovery following an immune system mediated attack on CNS myelin. As a result, an MBP-EAE animal model of MS was specifically used to identify the BDNF responsive cells during the early inflammatory phase of the disease process prior to de-myelination.
Materials and Methods
EAE Model & Neurological Testing
Experimental autoimmune encephalomyelitis (EAE) was induced using MBP, in adolescent female Lewis rats (Charles River, Montreal, QC) as previously described (Zhu et al., 2012). A total of 66 adolescent female Lewis rats were divided into 3 experimental groups: naïve control (NC), active control (AC) and experimental autoimmune encephalomyelitis (EAE). NC animals (n=6) did not receive any injections. AC animals (n=30) received 2 intraperitoneal injections (I.P.) ofpertussis toxin (PT; List Biological Laboratories CN: 180) (0.3 μg in 200 µl of phosphate buffered saline (PBS; Sigma: P-5368) at the identified time point’s day 0 & 2. In addition, these animals also received 2 x 50 μl s.c bilateral injections of Freund’s adjuvant (FA; (Sigma, CN: F-5506) + 500 μg of Mycobacterium Tuberculosis H37Ra (MT; Difco Laboratories, CN: 3114) + saline into the area above the base of the tail at day 0. EAE animals (n=30) received the same PT regimen administered to AC plus full inoculation with FA + 500 μg MT + 100 μg Guinea pig myelin basic protein (MBP; Cedarlane, CN: GP68-84 ) given as 2 x 50 μl s.c injections into the area above base of the tail. Animals in the AC and EAE groups were sacrificed for comparative protein and gene analysis at day 3, 6, 9, 12, and 15. Acronym identifier such as EAE3 refers to EAE animals – 3 days after inoculation vs AC3 refers to AC animals 3 days after inoculation and so forth. All animal experiments were conducted in accordance with the University of Manitoba Animal Users and Protocol Management Review Committee protocols, which comply with the Canadian Council on Animal Care guidelines. Our established experience with this model (Ben-Hur et al., 2007) indicates that EAE animals, begin to develop mild clinical symptoms by day 9 (tail weakness or paralysis) (Costa et al., 2003). By day 12 to 13 all animals experience a full range of neurological deficits such as tail and forelimb weakness, loss of bladder control and hind-limb paralysis. Based on our previous experience, with this model, the EAE rats enter into remission and regain motor function by 15 days post EAE induction. Female rats were specifically chosen because they are more predisposed to be affected than males (Orton et al., 2006). Neurological disability is scored according to the criteria detailed previously (Melanson et al., 2009). Neurological disability is scored according to the following criteria: Tail: 0 = normal; 1 = partially paralyzed, weakness; 2 = completely paralyzed, limp: Bladder: 0 = normal; 1 = incontinence: Right hind limb: 0 = normal; 1 = weakness; 2 = dragging with partial paralysis; 3 = complete paralysis: Left hind limb: 0 = normal; 1 = weakness; 2 = dragging with partial paralysis; 3 = complete paralysis: Right forelimb: 0 = normal; 1 = weakness; 2 = dragging, not able to support weight; 3 = complete paralysis: Left forelimb: 0 = normal; 1 = weakness; 2 = dragging, not able to support weight; 3 = complete paralysis. SC tissue will be collected at days 3, 6, 9, 12 and 15. Whole DNA/RNA and protein was collected using commercially available kits as described in our previous publications (Vora et al., 2010, Zhu et al., 2012, Begum et al., 2013). Changes in TrkB gene and protein expression levels will be assessed by standard in house protocols for immunohistochemistry (IHC), real time RT-PCR, and western blotting (WB) (Zhu et al., 2012, Vora et al., 2010, Melanson et al., 2009, Begum et al., 2013, Begum et al., 2010).
Real Time Reverse Transcription Polymerase Chain Reaction
Total RNA was isolated from SC tissue using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol. The mRNA was transcribed to cDNA and quantitative real time RT-PCR was conducted for measuring target gene as previously described (Zhu et al., 2012), using a Light-Cycler-DNA master SYBR green-1 kit following the manufacturer’s protocols (Bio-Rad, Hercules, CA, USA). TrkB primers were: forward: 5’-acgtcaccaatcacacggagtacc-3’; reverse; 5’-ctggcagagtcatcgtcgttgc-3’, annealing temperature 62°C. PCR product was calculated to a length of 430bp, as was confirmed on an ethidium bromide agarose gel. GADPH primers were: forward: 5’- aagaaggtggtgaagcaggcg-3’; reverse; 5’ — agacaacctggtcctcagtgtagc-3’.
Western Blot (WB)
Protein concentration for each sample was assessed using the Bradford protein assay. For each sample, 30 µg total protein was analyzed by WB as previously described (Zhu et al., 2012). Anti-TrkB antibody (1:500, R&D system Minneapolis, MN, USA) was used to detect BDNF protein. Following incubation with anti-rabbit secondary antibody (1:10,000, Jackson ImmunoResearch, West Grove, PA, USA), the enhanced chemiluminescence signal was detected by Multi Image ®II FluorChem®FC2 (Alpha Innotech, Santa Clara, CA, USA). Signals were normalized to GAPDH (1:1000; R&D Systems). Densitometry readings were performed using a FluorChem 8900 scanner (Alpha Innotech, San Leandro, CA, USA) with Alpha Ease FC software, and relative density ratio was calculated for the individual TrkB band density for each sample, vs. GAPDH levels. Three different cell preps were assayed by western blot and the mean densities for each band calculated, and their mean value was used (Frost et al., 2009).
Immunohistochemistry (IHC) Analysis
For IHC analysis of protein expression, animals were perfusion fixed with 4% paraformaldehyde as previously described (Zhu et al., 2012). Spinal columns were dissected free of overlying muscle and connective tissue, and decalcified according to previously described protocols (Begum et al., 2010). Qualitative IHC analysis of 10 µm cryostat sections were conducted according to previously described methods (Zhu et al., 2012). Double-labeled immunofluorescence using antibodies against MBP (1:50, Santa Cruz), GFAP (1:100, Santa Cruz), CD68 (ED1) (1:100, Santa Cruz), PDGFR-α(1:50, Santa Cruz), CD4 (1:100, Santa Cruz), and neuron specific β-III Tubulin (1:50, R&D) were conducted in conjunction with the polyclonal antibody TrkB (1:100, R&D system Minneapolis, MN, USA). Imaging was conducted using an Olympus BX51 configured with FV5000 confocal laser scanning capability. Acquired images were captured in Fluoview Version 4.3. Image sizing, black background balancing and final collation for publication were performed using Adobe Creative Suite 2 v9.0.2 (Adobe Systems Inc., San Jose, CA, USA). Cell size analysis was performed using Image Pro Express software (Media Cybernetics, Bethesda, MD, USA). No image manipulations were performed other than those described. Primary antibody and secondary antibody omission studies were also conducted on the samples(data not shown)
Axonal Diameter Size in the Dorsal Horn of the SC
The diameters of cross-sectional axons in SC tissue expressing TrkB were measured. TrkB positive (TrkB+) neurons were identified and measured in each of the 10 µm thick SC sections obtained from each experimental group (NC, AC, EAE). Imaging was performed using an Olympus IX81 scanning laser confocal microscope. Images were captured in Fluoview FV500 software. Image processing and nerve fiber diameters were measured (image of 100X magnification were used) using the Image Pro Express V6.0 (Media Cybernetics) software. To measure the nerve fiber diameters, a line was drawn from one edge of the axon to the opposite end to obtain a cross-sectional width of the nerve fibers. Measurements are expressed in microns. Statistical analysis of cell sizes were performed using GraphPad Prism version 4.03 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com. The data were analyzed using one way analysis of variance (ANOVA) with Bartlett’s test (for equal variances) and Dunnett’s Multiple Comparison Test. Significance was set at p<0.05. Significant differences between the means of different observational groups were determined.
Transmission Electron Microscopy (TEM)
SC ultrastructure was observed with TEM for changes in myelin protein structure and integrity. Animals from each experimental group (n =1) at day 12 post inoculation were perfused via left ventricle with a pre-fix solution containing 1I.U./ml heparin and 1% sodium nitrate in 0.9 % sodium chloride followed by a fixative solution of 4% of paraformaldehyde in Sorensen’s buffer (pH 7.3). Whole SCs were extracted and dissected into 1 mm by 1 mm sections and incubated by immersion with the same fixative solution for 3 hours, and then the solution was replaced with 5% sucrose in 0.1 M Sorensen’s buffer for 1 hour. Tissue sections were post-fixed using 1% of osmium tetroxide. The samples were dehydrated in ascendant alcohol and subsequently embedded in epon812 resin (E.M.S., Embed-812 embedding kit, 14120). Thin sections were stained with uranyl acetate and counterstained with lead citrate. SC ultrastructure was observed and micro photographed with a transmission electron microscope Philips CM10, at 60 kV, on ultra-thin sections (100 nm on 200 mesh grids) at the Histomorphology & Ultrastructural Imaging Unit of the University of Manitoba.
Statistical Analysis
Statistics was performed using GraphPad Prism version 5.04 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com. Results are expressed as mean ± SEM. The data were analyzed by one-way ANOVA with Tukey’s Multiple Comparison post hoc test. Student’s t-test was used to assess significance of differences between the groups. Normality and homogeneity of error variance of dependent variable was tested by using Kolmogorov-Smirnov and Levene’s test. A value of p<0.05 was considered significant.
Results
Neurological Disability
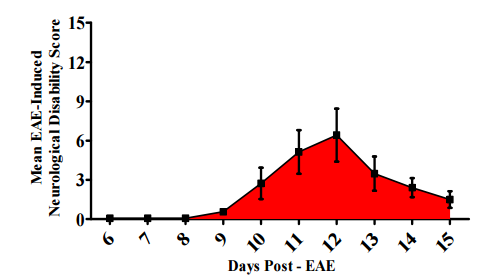
All animals in the EAE groups were assessed for neurological disability according to previously described protocols (Zhu et al., 2012, Zhu et al., 2013, Begum et al., 2013). None of the animals displayed neurological deficits prior to day EAE6 as evident by a score of zero. However, all EAE animals started to display signs of neurological disability by EAE9 representing the early inflammatory stage of disease induction (0.57 ± 0.45) (Figure 1). Mean peak neurological disability score (6.42 ± 5.35) was achieved by EAE12 which subsided by EAE15 (1.5 ± 1.41) as the animals entered the remission phase of disease. This pattern of neurological disability is well characterized for this animal model, and it has been consistent with our previous publication, give us the same graph published by (Begum et al., 2013) published in BioMed Research International. In contrast, NC and AC animals did not display neurological symptoms (data not shown).
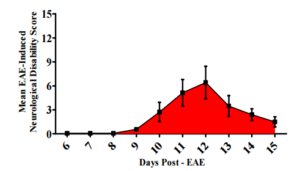
Figure 1: Neurological Disability Clinical Score for EAE Animals
This figure illustrates clinical scores in the EAE animals, at different times in the disease progression. Global neurological disability score obtained from assessment of all 6 specific clinical domains outlined in Materials and Methods (EAE model & Neurological Testing) Disability scores range from a mean clinical disability score of 0 (no disability) to 15 (maximal disability). Results indicate EAE animals developed signs of neurological disability at EAE 6 that peaked at EAE12 which subsided by EAE14 similarly to (Begum et al., 2013).
Expression of Trkb Protein in the EAE SC
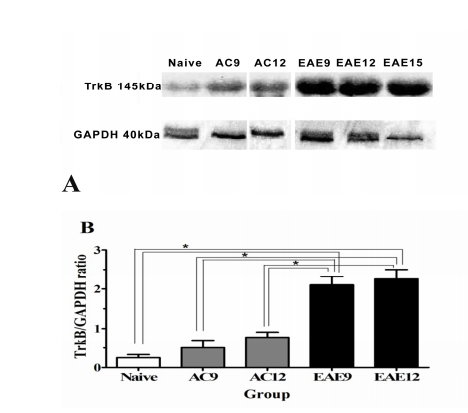
WB was used to determine the comparative time dependent TrkB protein expression amongst the 3 experimental groups (NC, AC and EAE). WB analysis revealed a significant increase in TrkB expression in EAE9 and EAE12 groups compared to NC and AC9 and AC12 groups (Figure 2A). Densitometry was conducted on the blots, and TrkB protein levels were assessed as a ratio of GAPDH levels. NC animals show TrkB protein expression at 0.235 ± 0.02. AC9 and AC12 animals show increased protein expression of TrkB relative to NCs (AC9 = 0.576 ± 0.12; AC12 = 0.807 ± 0.15). In comparison, EAE9 and EAE12 animals show a significant increase of TrkB expression in the SC over NCs and AC9 and AC12 (EAE9 = 2.124 ± 0.24 p<0.05 and EAE12 = 2.24 ± 0.26, p<0.05) (Figure 2B).
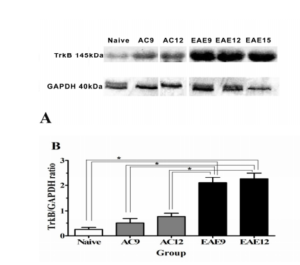
Figure 2: TrkB Protein & Gene Expression in the SC
Figure 2A: WB analysis of TrkB protein expression in the SC illustrates that full length TrkB (145kDa) protein is detected in naïve, active control and EAE groups at the different time points during disease progression. GAPDH protein was used as a loading control, and is detected at 40 kDa. (EAE9 = EAE animals 9 days post-inoculation; AC9 = 9 days post-inoculation; Naïve = NC animals)
Figure 2B: Densitometric analysis of WB for TrkB in SC shows increased TrkB protein expression in the SC of EAE animals (black bars) compared to NC (white bars) & AC (grey bars) SC, at different times in the disease progression. NC animals (white bars) show TrkB protein expression at 0.235 ± 0.02. AC9 & 12 animals [AC9 = 0.576 ± 0.12 and AC12 = 0.807 ± 0.15 (grey bars)] shown increased protein expression of TrkB compared to NCs. However, analysis of EAE animals (EAE9 = 2.124 ± 0.24 P<0.05 and EAE12 = 2.24 ± 0.26, P<0.05) show a significant increase of TrkB expression in the SC when comparatively assessed to NC (0.235 ± 0.02) and AC 9 &12 [AC9 = 0.576 ± 0.12 and AC12 = 0.807 ± 0.15].
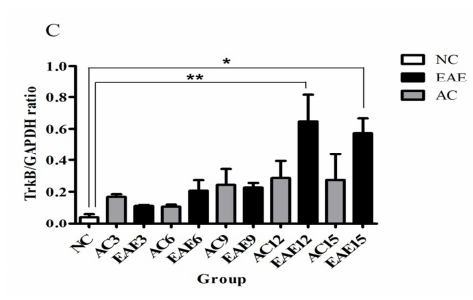
Expression of TrkB mRNA in the EAE SC
Real time RT-PCR was used to assess TrkB mRNA expression from all NC, AC and EAE groups. The data was normalized with GAPDH expression from each sample. Statistical analysis shows no significant difference in TrkB mRNA expression between EAE and AC groups (Figure 2C). However, EAE12 and EAE15 animals did show a significant increase in mRNA expression over NC group (p<0.01 and p<0.05 respectively) (* = p<0.05; ** = p<0.01; ANOVA followed by Tukey’s posthoc test).
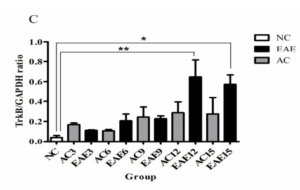
Figure 2C: This figure illustrates TrkB mRNA gene expression in SC at different times in the disease progression. Comparative Real time RT-PCR of TrkB expression in SC shows no significant difference in mRNA expression between the AC and EAE animal groups. However, EAE animals did show a significant increase in mRNA expression over NC group [NC = 0.0400 ± 0.0200] at days 12 and day 15 [EAE12 = 0.6467 ± 0.1683 EAE12 = 0.5733 ± 0.0933] (* = p<0.05; ** = p<0.01; ANOVA followed by Tukey’s posthoc test). Errors bars represent standard error of mean (SEM).
IHC Analysis of BDNF Responsive Cells in the SC Glial Cells
Double-labeled IHC staining was conducted to identify various types of BDNF responsive cells (TrkB expressing cells). A variety of cellular markers including: CD68 (macrophages, GFAP (astrocytes), PDGFRα (oligodendrocyte precursors) and MBP (mature OGs) were used to determine the expression of TrkB on glial cells. Our results show that TrkB was not co-localized with any of the other glial cell markers for which we have examined in the dorsal horn of the EAE SC (Data not shown).
Immune Cells
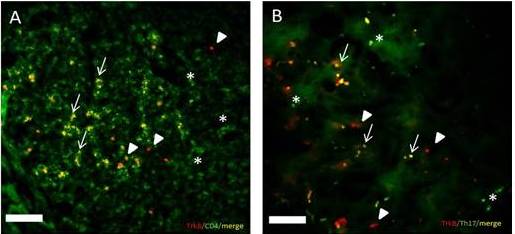
IHC analysis identified TrkB protein co-localization with CD4+ T-cells (Figure 3A) and Th17 cells in EAE SC (Figure 3B). These results confirm that immune cells express the TrkB receptor in EAE SC.
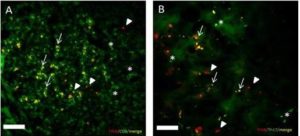
Figure 3: Trkb Expression by Immune Cells in the EAE SC
Figure 3A: TrkB and CD4 double labeling in EAE SC: This figure illustrates that IHC results (TrkB labeling red andCD4+ T cell labeling green) show TrkB is co-localized with CD4+ T cells (yellow). Images were taken at a total magnification of 100X from EAE 12 group. (Arrowheads depict TrkB labeling, * depict CD4+T cell labeling and arrows depict cells co-labeled for CD4+ T cells and TrkB). Bar = 20μm.
Figure 3B: TrkB and Th17 double labeling in EAE SC: This figure illustrates that IHC results (TrkB labeling red and Th17labeling green) show TrkB is co-localized with Th17 cells (yellow). Images were taken at a total magnification of 100X from EAE 12 group. (Arrowheads depict TrkB labeling,* depict Th17 labeling and Arrows depict co-labeling between TrkB and Th17 cells). Bar = 20μm.
Neuronal Cells
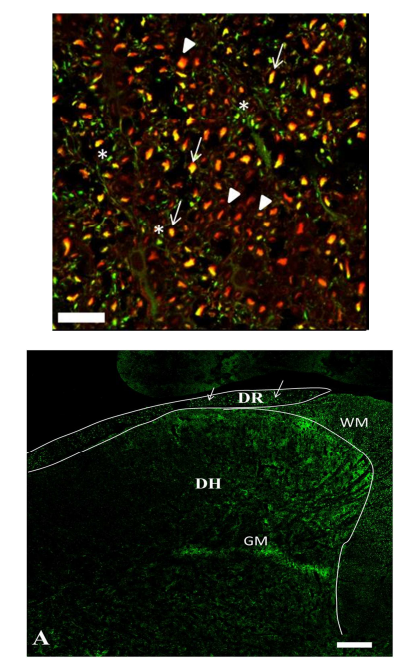
IHC analysis of EAE SC identified TrkB co-localization with the neuronal marker β-III-tubulin (Figure 4). Interestingly, IHC analysis also showed enhanced TrkB+ cells being localized specifically to the posterior marginalis and the nucleus proprius layers (Figure 5A) of the dorsal horn. IHC analysis suggests that the specific subsets of BDNF responsive neurons are likely to be Aδ fibers. The reason being is that Aδ fibers originate in the dorsal root ganglia and synapse in the posterior marginalis.
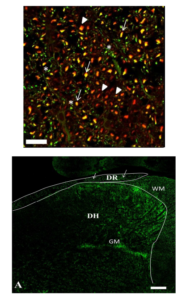
Figure 4: Trkb Expression by Neurons in the EAE SC
This figure illustrates that IHC results (TrkB labeling red (arrowheads) and β-III Tubulin labeling green (*) show TrkB is co-localized with neuronal axons (yellow- arrows). Images were taken at a total magnification of 100X from EAE 12 group. Bar = 20μm.
Measurement of Nerve Fibers Diameters in the Dorsal Horn of the SC
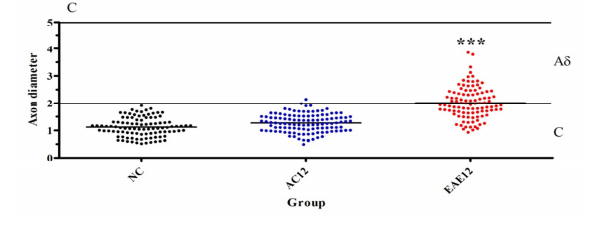
Dorsal horn nerve fibers expressing TrkB showed a statistically significant (p<0.0001) increase in size in the EAE12 group compared to AC animals at the same stage of the disease progression. NC SC nerve fibers had a mean diameter of 1.12 ± 0.34 μm, compared to AC animals with a mean diameter of 1.27 ± 0.33 μm. EAE animals have a mean nerve fiber diameter of 2.01 ± 0.58 μm which is highly significantly different from the AC nerve fiber diameters (*** = p<0.0001) – Figure 5B. These results suggest that the TrkB+ neuronal cell population switches from C fibers to Aδ fibers involvement following immune system activation. These findings correlate with the IHC findings described above, shown in Figure 5A.
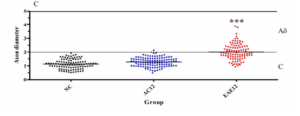
Figure 5: IHC Analysis of Trkb Positive Cells in the Dorsal Horn of the SC
Figure 5A – IHC staining of TrkB localization in the SC of EAE 12 show that BDNF responsive cells are located in theposterior marginalis at the dorsal root entry zone, and also in the nucleus proprius. Such cells correspond with Aδ fibers, which connect to the spinothalamic tract. IHC of TrkB in the SC of EAE12, shows TrkB (green spots) in the dorsal root(DR: white arrows indicate the dorsal root entry point) and grey matter (GM) of the SC. WM = white matter. Total magnification 10x bar = 100μm.
Figure 5B: Analysis of nerve fiber diameter of the TrkB positive neurons identified from SC nerve fibers in the dorsal horn. C fibers are on average 0.5-2.0 μm in diameter, while Aδ is on average 1-5μm in diameter. NC SC fibers have a mean diameter of 1.12 ± 0.34 μm, compared to AC12 with a mean diameter of 1.27 ± 0.33 μm. Interestingly the nerve fibers from EAE12 animals have a mean diameter of 2.01 ± 0.58 μm which is highly significantly different from the nerve fiber diameter for that of AC12 (*** = p<0.0001).
Myelin Pathology in the SC Observed in TEM
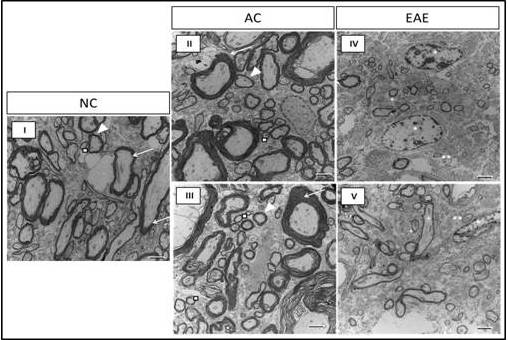
Since animals induced to a state of EAE exhibit peak neurological disability 12 days post-EAE induction, we aimed to assess the effect of EAE disease state on the structure and integrity of the myelin sheath. We believe changes in myelin structure and integrity contribute to the development of neurological disability seen in animals with EAE. We analyzed lumbar SC ultrastructure using TEM to assess the Comparative assessment of TEM images for NC, AC12, and EAE12 animals reveal significant myelin pathology during EAE (Figure 6 I-V). TEM images of the SCs of NC and AC12 animals reveal normal myelin ensheathment of various sized axons with no differences between the two groups (Figure 6 I, II, III). However, SC of EAE12 revealed the myelin sheath surrounding the axons of small to medium diameter neurons are markedly thinner than in both healthy and non-disease controls (Figure 6 IV, V). In addition, there is pronounced absence of myelin ensheathment in large diameter axons, as well as, the presence of necrotic cells. These changes in myelin integrity correlated with peak neurological disability (Figure 1).
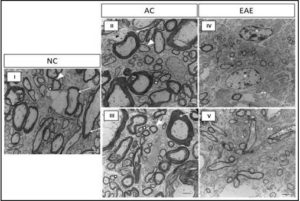
Figure 6: Changes in Myelin Protein Structure and Integrity Observed by TEM
Direct magnification 4600x using an AMT camera system. Scale bars at 2 µm. The TEM images captured in this figure were obtained from SC tissue subjected to identical experimental conditions and imaging procedures. I) SC tissue in NC shows normal myelin ensheathment of various sized axons; myelinated large diameter axons (white arrow), medium diameter (white arrow head), and small diameter axons (white square). II-III) There is normal myelin ensheathment in AC12 SC tissue; myelinated large diameter axons (white arrow), medium diameter (white arrow head), and small diameter axons (white square). IV-V) TEM images reveal myelin pathology in the SC tissue of EAE12 as demonstrated by the marked reduction of myelin ensheathment in small to medium diameter axons and an absence of myelin coating in large diameter axons. White** = small diameter non myelinated axons in EAE12 SC tissue. White + = thinly myelinated small to medium diameter axons in EAE12 SC tissue.
Discussion
Although the exact mechanisms underlying MS remain unknown, several therapeutic candidate molecules have been suggested to have a significant beneficial role in repairing damaged myelin and reversing the neurological disabilities associated with this white matter disorder (Namaka et al., 2011a, Melanson et al., 2009). Specifically, recent studies have identified BDNF as a critical regulator of myelin repair following immune system mediated destruction of CNS myelin (Zhu et al., 2012, VonDran et al., 2011, Cosgaya et al., 2002, Lee et al., 2012). However, there is little understanding of the exact role of BDNF in MS. We hypothesized that the TrkB receptor for BDNF represents a critical component essential to the beneficial molecular signaling of BDNF that is involved in myelin repair following EAE induced myelin damage. Therefore, we designed a study focused on assessing the time dependent expression changes of the high-affinity receptor TrkB in the SC following EAE induction.
Our data demonstrates significant increases in the protein expression for TrkB in the SC of EAE compared to NC animals and AC animals at the same time points following EAE induction. Interestingly, based on the SC distribution of the TrkB+ cells within the dorsal horn (Figure 5A), we show that these BDNF responsive cells correspond to the anatomical location and nerve fiber diameter of those neurons involved in the transmission of pain. Based on our earlier published EAE studies (Zhu et al., 2012), we demonstrated that BDNF is up regulated at day 12 in the dorsal horn of the SC following EAE induction (Zhu et al., 2012). Interestingly BDNF signaling via TrkB in the dorsal horn of the SC is known to contribute to neuropathic pain (NPP) by activation of microglia (Zhou et al., 2011). NPP is a chronic syndrome commonly suffered by patients with MS (Chatel et al., 2001). It is proposed NPP and the early induction of MS share molecular mechanisms (Truini et al., 2013). As such, up to 50% of MS patients present with NPP prior to their diagnosis or in the earliest stages of disease progression (Solaro et al., 2013, Osterberg and Boivie, 2009, Svendsen et al., 2003), which suggests the development of NPP in patients with pre-disposing risk factors for MS may serve as a pre-diagnostic indicator for MS. Therefore, the up regulation of BDNF (Zhu et al., 2012) and increase in cellular responsiveness (via TrkB) to local BDNF release in the dorsal horn SC may contribute to the development of NPP. Thus, an elevation in BDNF and TrkB may likewise serve as early indicators for the development of MS.
In this study we showed a significant up-regulation in TrkB protein expression (EAE 9 – 15) throughout theSC which suggests increased cellular responsiveness to BDNF via TrkB thereby contributing to neurological recovery. This is consistent with the findings of Song et al , which showed that reduced TrkB expression increased EAE disease severity (Song et al., 2013). Interestingly, our IHC data showed the absence of TrkB expression in SC astrocytes during EAE. However, recent SC injury studies indicate that the TrkB receptor on astrocytes sequester the incoming BDNF protein in injured SC (Liebl et al., 2001, Zhang et al., 2012), thereby preventing BDNF from exerting its beneficial effects on re-myelination and/or myelin repair. Our data in this regard, suggests that BDNF sequestering may not be possible due to the absence of astrocyte TrkB expression. Thus, this would allow DRG-derived BDNF to promote neurological recovery/remission and exert its effects on myelin repair following an EAE induced insult to CNS myelin. BDNF signaling via TrkB has been shown to be protective, reducing disease severity during the early mild phase of EAE (Song et al., 2013). Our results using an MBP inflammatory model of EAE also support these findings. Interestingly, this is not the case during severe disease (Song et al., 2008). In fact, it has been demonstrated that an increase in TrkB expression in astrocytes in MS and EAE lesions facilitates nitric oxide production and neurodegeneration that inhibit re-myelination (Colombo et al., 2012, Stadelmann et al., 2002, Aharoni et al., 2005, De Santi et al., 2009). As such, researchers must be aware of the differential effects that correspond to the differential progression stages of MS. Hence, these later findngs suggest that in the later stages of disease progression TrkB signaling may modulate disease outcome and severity. Thus differential TrkB signaling may lead to different biological outcomes that are dependent on time throughout disease course. Accordingly, it is not surprising then that during an early inflammatory pre-demyelinating model of EAE we noted an absence of astrocyte TrkB expression. This is likely because the duration of the study was too short to demonstrate long term disease progression. Henceforth, the lack of TrkB containing astrocytes during early EAE induction may help account for recovery from neurological disability by EAE15. As a result, our study identifies that the early inflammatory stages following EAE may represent the most optimal time for targeted treatment strategies to reverse the downstream effects of myelin damage that are known to occur as a function of time. However, additional studies are required to confirm this.
As a result, there appears to be a narrow window of time in the early stages of MS by which BDNF can beneficially signal through TrkB to promote myelin repair. As the disease progresses, there appears to be a continued elevation of TrkB containing astrocytes. These TrkB+ astrocytes eventually take over and prevent complete myelin repair at the lesioned site by sequestering BDNF. As a result, patients in the later stages of disease progression are left with a permanent neurological disability for which they can’t recover. Based on the current knowledge of MS, patients with relapsing remitting MS (RRMS) eventually develop secondary progressive MS (SPMS). SPMS is the clinically recognized progressive phase of the disease where patients experience a continued decline of neurological function without any periods of remission/myelin repair. The TrkB findings presented from our research in the early inflammatory stages of EAE taken together with the findings of other EAE researchers involving the later stages of the disease, confirm the importance of BDNF and TrkB in myelin repair. Our research also suggests the importance of astrocytes in preventing myelin repair in the later stages of MS.
Interestingly, in our study we did not see TrkB co-localized with oligodendrocyte immunoreactivity. This contradicts previously published studies, which have shown SC OGs express TrkB (McCartney et al., 2008). The difference between our study and the previous studies may be as a result of the use of different antibodies to identify cells of the oligodendrocyte lineage, or the type of animal model being studied.
Furthermore, we did not identify TrkB+ cells in NC SC grey matter, which correlates with one previous study (McCartney et al., 2008). However, it is possible that TrkB expression in OGs does not occur in the early stages of the disease progression, further studies of the later secondary de-myelinating phase of the disease may reveal a role for BDNF in the direct regulation of re-myelinating events. Alternatively, BDNF may regulate OGs via pro-BDNF via p75NTR interactions on OGs. ProBDNF is known to promote myelination via p75NTR-mediated activity independent of TrkB (Cosgaya et al., 2002). BDNF binds to two neurotrophin receptors, p75NTR and TrkB (Schecterson and Bothwell, 2010). Pro-BDNF preferentially binds to p75NTR (Chao, 1994), and the mature form of BDNF binds with high affinity to the TrkB receptor (Arevalo and Wu, 2006). Studies show that OGs express p75NTR , expression of which is down regulated after axonal contact is made (Cohen et al., 1996). Thus, further studies are required to identify the differential expression of p75NTR and TrkB in OG lineage cells.
Several studies have shown that CD4+ cells are an important source of BDNF that contributes to immune mediated neuroprotection (Linker et al., 2010). Our results show that cells of immune origin specifically CD4+ Th cells and Th17 cells, also express TrkB and are therefore responsive to BDNF signaling. BDNF appears to regulate the differentiation of thymocytes to their CD4+ or CD8+ mature status (Maroder et al., 1996). Thus it is plausible that in the early stages of EAE disease progression (as assessed in our study), the TrkB+ CD4+ cells we identified are recently differentiated Th cells. These cells may lose their BDNF responsiveness as they mature. Th17 cells have recently been identified as playing a critical role in the MS disease process (El-Behi et al., 2010). However, there have been no studies investigating the role of BDNF in the regulation of Th17 cells. It is possible that their differentiation is also regulated by BDNF in the early stages of maturation. It is also possible that BDNF acts to protect the Th17 cells from activation-induced apoptosis (De Santi et al., 2011). Further studies are required to confirm a role for BDNF in regulating Th17 cells. Inflammatory responses can provide regenerative and protective effects during CNS damage (Moalem et al., 1999). Thus the TrkB+ T-cells could play an important role in MS immunopathogenesis by modulating auto-reactive T-cells.
Our study also shows that TrkB immunoreactivity is localized specifically to the axons of SC in the EAE12 experimental group. This localization of TrkB in the SC axons may represent the earliest stages by which any axonal transport of BDNF from the DRG may be sequestered thereby preventing it from reaching its target site at the MS lesion.Interestingly, qRT-PCR data show that there is no significant change in TrkB mRNA expression between EAE and AC groups. These results suggest that increased SC TrkB protein expression may be as a result of increased translation in the axons. Our previous study showed that the predominant source of BDNF protein expression in the DRG is small diameter (1-5 mm) Aδ sensory nerve fibers (Zhu et al., 2012). Consistently, our current study identifies Aδ fibers as the predominant TrkB+ (BDNF responsive) cells in the dorsal horn. Additionally, while large diameter axons are absent of myelin, small to medium sized axons exhibit only a marked reduction of myelin ensheathment based on TEM analysis. Although, a marked thinning of myelin coating in small to medium diameter neurons may compromise the structural integrity of Aδ fibers, BDNF transport mechanisms may likely still be functional during EAE (Zhu et al., 2012). Further, our results suggest that the TrkB+ neuronal cell population switches from C fibers to Aδ fibers following immune system activation, which may serve to increase BDNF transport from the DRG to SC. As a result, the increase expression of TrkB in Aδ fibers and the presence of thinly myelinated small to medium axons support our hypothesis that the transport of DRG-derived BDNF into the SC via the DRG-SC connective pathway is critical for neurological recovery and myelin repair.
Sensory neurons are classified according to conduction velocity, which easily translates to axonal diameter. The smallest fibers, C and Aδ fibers, are pain and temperature receptors, and synapse in different layers of the dorsal horn. The three main layers of the dorsal horn are the posterior marginalis, the substantia gelatinosa, and the nucleus proprius. Aδ pain fibers enter the posterior marginalis and the nucleus proprius, and synapse with sensory afferent neurons that carry pain signals to the thalamus via the spinothalamic tract. The spinothalamic system is one of the major pain-transmission pathways (Willis et al., 1999). The C fibers enter the substantia gelatinosa and synapse on interneurons, which do not project out of the immediate area. Thus our data showing that EAE SC has a higher proportion of Aδ fibers suggests that the mode of BDNF induced pain transmission is altered during the course of the disease. In addition, the expression of TrkB in the nucleus proprius shows that the Aδ fibers are signaling directly with sensory afferents associated with the spinothalamic tract. This finding correlates with our studies showing that EAE animals are more sensitive to cold than active control animals at the same phase of the disease (Zhu et al., 2013).
Conclusion
Our study provides evidence of BDNF responsive cells in the SC in an animal model of MS, which support the role of TrkB in modulating acute inflammatory processes that contribute to myelin damage and associated neurological disability. We observed an up-regulation of TrkB in SC at day 12 post-EAE induction, while Zhu et al. in 2012 showed an up-regulation of BDNF in SC at the same time point. Both these findings correspond to the inflammatory peak of the acute phase of EAE. Further, we revealed TrkB expression is up-regulated in several SC cell types including Aδ nerve fibres but absent in astrocytes. Finally, we showed myelin ensheathment is compromised in various size axons in an EAE model normally associated with little to no demyelination.
Other studies show BDNF is protective during the early mild phase of EAE but not during severe disease. This may be explained by astrocyte sequestering of BDNF via TrkB which prevents BDNF from exerting its beneficial effects. Since our study does not show TrkB expression in astrocytes, we suggest the TrkB receptor may be a disease modulator dependent on disease time course and cell type. Therefore, this suggests a narrow window may exist during the early inflammatory phase of EAE when a precise balance between ligand (BDNF) and receptor (TrkB) is achieved to promote neurological recovery. Therefore, this may provide insights into the optimal time for early treatment in MS patients. Further studies are also required to study the expression of p75NTR receptor in EAE SC to determine the effect of TrkB independent BDNF activity.
Acknowledgements
We would like to thank our funding & supporting agencies: Canadian Paraplegic Association (CPA), Pfizer UK Global (MN), and Pfizer Canada (MN). WZ was the recipient of a Canada Millennium Scholarship Foundation award. Manitoba Multiple Sclerosis Research Network (MMSRN).
Statement of interest
The authors confirm that there are no conflicts of interest.
References
Acosta, C. M. R., Cortes, C., Macphee, H. & Namaka, M. P. (2013). “Exploring the Role of Nerve Growth Factor in Multiple Sclerosis: Implications in Myelin Repair,” CNS & Neurological Disorders – Drug Targets, 12 (8) 1242-1256
Publisher – Google Scholar
Aharoni, R., Eilam, R., Domev, H., Labunskay, G., Sela, M. & Arnon, R. (2005). “The Immunomodulator Glatiramer Acetate Augments the Expression of Neurotrophic Factors in Brains of Experimental Autoimmune Encephalomyelitis Mice,” Proceedings of the National Academy of Sciences of the United States of America, 102 (52) 19045-50
Publisher – Google Scholar
Althaus, H. H., Kloppner, S., Klopfleisch, S. & Schmitz, M. (2008). “Oligodendroglial Cells and Neurotrophins: A Polyphonic Cantata in Major and Minor,” Journal of Molecular Neuroscience, 35 (1) 65-79
Publisher – Google Scholar
Arevalo, J. C. & Wu, S. H. (2006). “Neurotrophin Signaling: Many Exciting Surprises!,” Cellular and Molecular Life Sciences CMLS, 63 (13) 1523-37
Publisher – Google Scholar
Armstrong, R. C., Le, T. Q., Frost, E. E., Borke, R. C. & Vana, A. C. (2002). “Absence of Fibroblast Growth Factor 2 Promotes Oligodendroglial Repopulation of Demyelinated White Matter,” The Journal of Neuroscience, 22 (19) 8574-85
Publisher – Google Scholar
Begum, F., Zhu, W., Cortes, C., MacNeil, B. & Namaka, M. (2013). “Elevation of Tumor Necrosis Factor Alpha in Dorsal Root Ganglia and Spinal Cord Is Associated with Neuroimmune Modulation of Pain in an Animal Model of Multiple Sclerosis,” Journal of Neuroimmune Pharmacology, 8 (3) 677-90
Publisher – Google Scholar
Begum, F., Zhu, W., Namaka, M. P. & Frost, E. E. (2010). “A Novel Decalcification Method for Adult Rodent Bone for Histological Analysis of Peripheral-Central Nervous System Connections,” Journal of Neuroscience Methods, 187 (1) 59-66
Publisher – Google Scholar
Ben-Hur, T., Van Heeswijk, R. B., Einstein, O., Aharonowiz, M., Xue, R., Frost, E. E., Mori, S., Reubinoff, B. E. & Bulte, J. W. (2007). “Serial in Vivo MR Tracking of Magnetically Labeled Neural Spheres Transplanted in Chronic EAE Mice,” Magnetic Resonance in Medicine, 57 (1) 164-71
Publisher – Google Scholar
Bibel, M., Hoppe, E. & Barde, Y. A. (1999). “Biochemical and Functional Interactions between the Neurotrophin Receptors Trk and p75NTR,” The EMBO Journal, 18 (3) 616-22
Publisher – Google Scholar
Binder, D. K. & Scharfman, H. E. (2004). “Brain-Derived Neurotrophic Factor,” Growth Factors, 22 (3) 123-31
Publisher – Google Scholar
Cellerino, A., Carroll, P., Thoenen, H. & Barde, Y. A. (1997). “Reduced Size of Retinal Ganglion Cell Axons and Hypomyelination in Mice Lacking Brain-Derived Neurotrophic Factor,” Molecular and Cellular Neuroscience, 9 (5-6) 397-408
Publisher – Google Scholar
Chao, M. V. (1994). “The p75 Neurotrophin Receptor,” Journal of Neurobiology, 25 (11) 1373-85
Publisher – Google Scholar
Chatel, M., Lanteri-Minet, M. & Lebrun-Frenay, C. (2001). “[Pain in Multiple Sclerosis],” Revue Neurologique (Paris), 157 (8-9 Pt 2) 1072-8
Publisher – Google Scholar
Cohen, R. I., Marmur, R., Norton, W. T., Mehler, M. F. & Kessler, J. A. (1996). “Nerve Growth Factor and Neurotrophin-3 Differentially Regulate the Proliferation and Survival of Developing Rat Brain Oligodendrocytes,” The Journal of Neuroscience, 16 (20) 6433-6442
Publisher – Google Scholar
Colombo, E., Cordiglieri, C., Melli, G., Newcombe, J., Krumbholz, M., Parada, L. F., Medico, E., Hohlfeld, R., Meinl, E. & Farina, C. (2012). “Stimulation of the Neurotrophin Receptor TrKB on Astrocytes Drives Nitric Oxide Production and Neurodegeneration,” The Journal of Experimental Medicine, 209 (3) 521-35
Publisher – Google Scholar
Cosgaya, J. M., Chan, J. R. & Shooter, E. M. (2002). “The Neurotrophin Receptor P75ntr as a Positive Modulator of Myelination,” Science, 298 (5596) 1245-8
Publisher – Google Scholar
Costa, O., Divoux, D., Ischenko, A., Tron, F. & Fontaine, M. (2003). “Optimization of an Animal Model of Experimental Autoimmune Encephalomyelitis Achieved with a Multiple MOG (35-55) Peptide in C57BL6/J Strain of Mice,” Journal of Autoimmunity, 20 (1) 51-61
Publisher – Google Scholar
De Santi, L., Annunziata, P., Sessa, E. & Bramanti, P. (2009). “Brain-Derived Neurotrophic Factor and Trkb Receptor in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis,” Journal of the Neurological Sciences, 287 (1-2) 17-26
Publisher – Google Scholar
De Santi, L., Polimeni, G., Cuzzocrea, S., Esposito, E., Sessa, E., Annunziata, P. & Bramanti, P. (2011). “Neuroinflammation and Neuroprotection: An Update on (Future) Neurotrophin-Related Strategies in Multiple Sclerosis Treatment,” Current Medicinal Chemistry, 18 (12) 1775-84
Publisher – Google Scholar
Djalali, S., Holtje, M., Grosse, G., Rothe, T., Stroh, T., Grosse, J., Deng, D. R., Hellweg, R., Grantyn, R., Hortnagl, H. & Ahnert-Hilger, G. (2005). “Effects of Brain-Derived Neurotrophic Factor (BDNF) on Glial Cells and Serotonergic Neurones during Development,” Journal of Neurochemistry, 92 (3) 616-627
Publisher – Google Scholar
Du, Y., Lercher, L. D., Zhou, R. & Dreyfus, C. F. (2006). “Mitogen-Activated Protein Kinase Pathway Mediates Effects of Brain-Derived Neurotrophic Factor on Differentiation of Basal Forebrain Oligodendrocytes,” Journal of Neuroscience Research, 84 (8) 1692-702
Publisher – Google Scholar
El-Behi, M., Rostami, A. & Ciric, B. (2010). “Current Views on the Roles of Th1 and Th17 Cells in Experimental Autoimmune Encephalomyelitis,” Journal of Neuroimmune Pharmacology, 5 (2) 189-98
Publisher – Google Scholar
Emery, B. (2011). “Regulation of Oligodendrocyte Differentiation and Myelination,” Science, 330 (6005) 779-82
Publisher – Google Scholar
Emery, E., Aldana, P., Bunge, M. B., Puckett, W., Srinivasan, A., Keane, R. W., Bethea, J. & Levi, A. D. (1998). “Apoptosis after Traumatic Human Spinal Cord Injury,” Journal of Neurosurgery, 89 (6) 911-20
Publisher – Google Scholar
Frost, E. E., Nielsen, J. A., Le, T. Q. & Armstrong, R. C. (2003). “PDGF and FGF2 Regulate Oligodendrocyte Progenitor Responses to Demyelination,” Journal of Neurobiology, 54 (3) 457-72
Publisher – Google Scholar
Frost, E. E., Zhou, Z., Krasnesky, K. & Armstrong, R. C. (2009). “Initiation of Oligodendrocyte Progenitor Cell Migration by a PDGF-A Activated Extracellular Regulated Kinase (ERK) Signaling Pathway,” Neurochemical Research, 34 (1) 169-181
Publisher – Google Scholar
Genain, C. P. & Hauser, S. L. (1996). “Allergic Encephalomyelitis in Common Marmosets: Pathogenesis of a Multiple Sclerosis-Like Lesion,” Methods in Enzymology, 10 (3) 420-34
Publisher – Google Scholar
Gensert, J. A. M. & Goldman, J. E. (1997). “Endogenous Progenitors Remyelinate Demyelinated Axons in the Adult CNS,” Neuron, 19 (1) 197-203
Publisher – Google Scholar
Gogate, N., Verma, L., Zhou, J. M., Milward, E., Rusten, R., O’connor, M., Kufta, C., Kim, J., Hudson, L. & Dubois-Dalcq, M. (1994). “Plasticity in the Adult Human Oligodendrocyte Lineage,” The Journal of Neuroscience, 14 (8) 4571-87
Publisher – Google Scholar
Horner, P. J., Power, A. E., Kempermann, G., Kuhn, H. G., Palmer, T. D., Winkler, J., Thal, L. J. & Gage, F. H. (2000). “Proliferation and Differentiation of Progenitor Cells throughout the Intact Adult Rat Spinal Cord,” The Journal of Neuroscience, 20 (6) 2218-28
Publisher – Google Scholar
Ishii, K., Toda, M., Nakai, Y., Asou, H., Watanabe, M., Nakamura, M., Yato, Y., Fujimura, Y., Kawakami, Y., Toyama, Y. & Uyemura, K. (2001). “Increase of Oligodendrocyte Progenitor Cells after Spinal Cord Injury,” Journal of Neuroscience Research, 65 (6) 500-7
Publisher – Google Scholar
Keirstead, H. S., Levine, J. M. & Blakemore, W. F. (1998). “Response of the Oligodendrocyte Progenitor Cell Population (Defined By NG2 Labelling) To Demyelination of the Adult Spinal Cord,” Glia, 22 (2) 161-70
Publisher – Google Scholar
Kolbeck, R., Jungbluth, S. & Barde, Y. A. (1994). “Characterisation of Neurotrophin Dimers and Monomers,” European Journal of Biochemistry, 225 (3) 995-1003
Publisher – Google Scholar
Lee, D. H., Geyer, E., Flach, A. C., Jung, K., Gold, R., Flugel, A., Linker, R. A. & Luhder, F. (2012). “Central Nervous System Rather than Immune Cell-Derived BDNF Mediates Axonal Protective Effects Early in Autoimmune Demyelination,” Acta Neuropathologica, 123 (2) 247-58
Publisher – Google Scholar
Lee, F. S., Kim, A. H., Khursigara, G. & Chao, M. V. (2001). “The Uniqueness of Being a Neurotrophin Receptor,”Current Opinion in Neurobiology, 11 (3) 281-6
Publisher – Google Scholar
Liebl, D. J., Huang, W., Young, W. & Parada, L. F. (2001). “Regulation of Trk Receptors Following Contusion of the Rat Spinal Cord,” Experimental Neurology, 167 (1) 15-26
Publisher – Google Scholar
Linker, R., Gold, R. & Luhder, F. (2009). “Function of Neurotrophic Factors beyond the Nervous System: Inflammation and Autoimmune Demyelination,” Critical Reviews™ in Immunology, 29 (1) 43-68
Publisher – Google Scholar
Linker, R. A., Lee, D. H., Demir, S., Wiese, S., Kruse, N., Siglienti, I., Gerhardt, E., Neumann, H., Sendtner, M., Luhder, F. & Gold, R. (2010). “Functional Role of Brain-Derived Neurotrophic Factor in Neuroprotective Autoimmunity: Therapeutic Implications in a Model of Multiple Sclerosis,” Brain, 133 (Pt 8) 2248-63
Publisher – Google Scholar
Lu, J., Kurejova, M., Wirotanseng, L. N., Linker, R. A., Kuner, R. & Tappe-Theodor, A. (2012). “Pain in Experimental Autoimmune Encephalitis: A Comparative Study between Different Mouse Models,” Journal of Neuroinflammatio, 9 (1) 233
Publisher – Google Scholar
Lykissas, M. G., Batistatou, A. K., Charalabopoulos, K. A. & Beris, A. E. (2007). “The Role of Neurotrophins in Axonal Growth, Guidance, and Regeneration,” Current Neurovascular Research, 4 (2) 143-51
Publisher – Google Scholar
Lytle, J. M., Chittajallu, R., Wrathall, J. R. & Gallo, V. (2009). “NG2 Cell Response in the CNP-EGFP Mouse after Contusive Spinal Cord Injury,” Glia, 57 (3) 270-85
Publisher – Google Scholar
Maroder, M., Bellavia, D., Meco, D., Napolitano, M., Stigliano, A., Alesse, E., Vacca, A., Giannini, G., Frati, L., Gulino, A. & Screpanti, I. (1996). “Expression of Trkb Neurotrophin Receptor during T Cell Development. Role of Brain Derived Neurotrophic Factor in Immature Thymocyte Survival,” The Journal of Immunology, 157 (7) 2864-72
Publisher – Google Scholar
Mccartney, A. M., Abejuela, V. L. & Isaacson, L. G. (2008). “Characterization of Trkb Immunoreactive Cells in the Intermediolateral Cell Column of the Rat Spinal Cord,” Neuroscience Letters, 440 (2) 103-8
Publisher – Google Scholar
McTigue, D. M., Wei, P. & Stokes, B. T. (2001). “Proliferation of NG2-Positive Cells and Altered Oligodendrocyte Numbers in the Contused Rat Spinal Cord,” The Journal of Neuroscience, 21 (10) 3392-400
Publisher – Google Scholar
Mekhail, M., Almazan, G. & Tabrizian, M. (2012). “Oligodendrocyte-Protection and Remyelination Post-Spinal Cord Injuries: A Review,” Progress in Neurobiology, 96, (3), 322—339
Publisher – Google Scholar
Melanson, M., Miao, P., Eisenstat, D., Gong, Y., Gu, X., Au, K., Zhu, W., Begum, F., Frost, E. & Namaka, M. (2009). “Experimental Autoimmune Encephalomyelitis-Induced Upregulation of Tumor Necrosis Factor-Alpha in the Dorsal Root Ganglia,” Multiple Sclerosis, 15 (10) 1135-1145
Publisher – Google Scholar
Messersmith, D. J., Murtie, J. C., Le, T. Q., Frost, E. E. & Armstrong, R. C. (2000). “Fibroblast Growth Factor 2 (FGF2) and FGF Receptor Expression in an Experimental Demyelinating Disease with Extensive Remyelination,” Journal of Neuroscience Research, 62 (2) 241-56
Publisher – Google Scholar
Moalem, G., Leibowitz-Amit, R., Yoles, E., Mor, F., Cohen, I. R. & Schwartz, M. (1999). “Autoimmune T Cells Protect Neurons from Secondary Degeneration after Central Nervous System Axotomy,” Nature Medicine, 5 (1) 49-55
Publisher – Google Scholar
Nakajima, H., Uchida, K., Yayama, T., Kobayashi, S., Guerrero, A. R., Furukawa, S. & Baba, H. (2010). “Targeted Retrograde Gene Delivery of Brain-Derived Neurotrophic Factor Suppresses Apoptosis of Neurons and Oligodendroglia after Spinal Cord Injury in Rats,” Spine (Phila Pa 1976), 35 (5) 497-504
Publisher – Google Scholar
Namaka, M., Kapoor, S., Simms, L., Leong, C., Grossberndt, A., Prout, M., Frost, E., Esfahani, F., Gomori, A. & Mulvey, M. R. (2011a). “Molecular Mimicry and Multiple Sclerosis,” Neural Regeneration Research, 6 (17) 1322-1333
Publisher – Google Scholar
Namaka, M. P., Ethans, K., Jensen, B., Esfahani, F. & Frost, E. E. (2011b). ‘Multiple Sclerosis. Therapeutic Choices/E-Therapeutic,’ Ottawa: Canadian Pharmacists Association Publishers, Inc.
Orton, S. M., Herrera, B. M., Yee, I. M., Valdar, W., Ramagopalan, S. V., Sadovnick, A. D. & Ebers, G. C. (2006). “Sex Ratio of Multiple Sclerosis in Canada: A Longitudinal Study,” The Lancet Neurology, 5 (11) 932-6
Publisher – Google Scholar
Osterberg, A. & Boivie, J. (2010). “Central Pain in Multiple Sclerosis – Sensory Abnormalities,” European Journal of Pain, 14 (1) 104—110
Publisher – Google Scholar
Pang, P. T., Teng, H. K., Zaitsev, E., Woo, N. T., Sakata, K., Zhen, S., Teng, K. K., Yung, W. H., Hempstead, B. L. & Lu, B. (2004). “Cleavage of ProBDNF by tPA/Plasmin is Essential For Long-Term Hippocampal Plasticity,” Science, 306 (5695) 487-91
Publisher – Google Scholar
Patapoutian, A. & Reichardt, L. F. (2001). “Trk Receptors: Mediators of Neurotrophin Action,” Current Opinion in Neurobiology, 11 (3) 272-80
Publisher – Google Scholar
Schecterson, L. C. & Bothwell, M. (2010). “Neurotrophin Receptors: Old Friends with New Partners,” Developmental Neurobiology, 70 (5) 332-8
Publisher – Google Scholar
Seidah, N. G., Benjannet, S., Pareek, S., Chretien, M. & Murphy, R. A. (1996). “Cellular Processing of the Neurotrophin Precursors of NT3 and BDNF by the Mammalian Proprotein Convertases,” FEBS Letters, 379 (3) 247-50
Publisher – Google Scholar
Shamovsky, I. L., Ross, G. M., Riopelle, R. J. & Weaver, D. F. (1999). “The Interaction of Neurotrophins with the P75NTR Common Neurotrophin Receptor: A Comprehensive Molecular Modeling Study,” Protein Science, 8 (11) 2223-33
Publisher – Google Scholar
Solaro, C., Trabucco, E. & Messmer Uccelli, M. (2013). “Pain and Multiple Sclerosis: Pathophysiology and Treatment,”Current Neurology and Neuroscience Reports, 13 (1) 320
Publisher – Google Scholar
Song, F., Bandara, M., Deol, H., Loeb, J. A., Benjamins, J. & Lisak, R. P. (2013). “Complexity of Trophic Factor Signaling in Experimental Autoimmune Encephalomyelitis: Differential Expression of Neurotrophic and Gliotrophic Factors,” Journal of Neuroimmunology, 262 (1-2) 11-8
Publisher – Google Scholar
Song, X. Y., Li, F., Zhang, F. H., Zhong, J. H. & Zhou, X. F. (2008). “Peripherally-Derived BDNF Promotes Regeneration of Ascending Sensory Neurons after Spinal Cord Injury,” PloS One, 3 (3) E1707
Publisher – Google Scholar
Stadelmann, C., Kerschenstainer, M., Misgeld, T., Bruck, R., Hohlfeld, R. & Lassmann, H. (2002). “BDNF and Gp145trkb in Multiple Sclerosis Brain Lesions: Neuroprotective Interactions between Immune and Neuronal Cells?,” Brain, 125 (1) 75-85
Publisher – Google Scholar
Stepanov, A. V., Belogurov, A. A., Mamedov, A. E., Melamed, D., Smirnov, I. V., Kuzina, E. S., Genkin, D. D., Boiko, A. N., Sharanova, S. N., Bacon, A., Ponomarenko, N. A. & Gabibov, A. G. (2012). “[Therapeutic Effect of Encapsulated into the Nanocontainers MBP Immunodominant Peptides on EAE Development in DA Rats],” Bioorganičeskaâ Himiâ, 38 (3) 306-14
Publisher – Google Scholar
Svendsen, K. B., Jensen, T. S., Overvad, K., Hansen, H. J., Koch-Henriksen, N. & Bach, F. W. (2003). “Pain in Patients with Multiple Sclerosis: A Population-Based Study,” Archives of Neurology, 60 (8) 1089-1094
Publisher – Google Scholar
Teng, H. K., Teng, K. K., Lee, R., Wright, S., Tevar, S., Almeida, R. D., Kermani, P., Torkin, R., Chen, Z. Y., Lee, F. S., Kraemer, R. T., Nykjaer, A. & Hempstead, B. L. (2005). “ProBDNFInduces Neuronal Apoptosis via Activation of a Receptor Complex of p75NTR and Sortilin,” The Journal of Neuroscience, 25 (22) 5455-63
Publisher – Google Scholar
Totoiu, M. O. & Keirstead, H. S. (2005). “Spinal Cord Injury is Accompanied by Chronic Progressive Demyelination,”Journal of Comparative Neurology, 486 (4) 373-83
Publisher – Google Scholar
Truini, A., Barbanti, P., Pozzilli, C. & Cruccu, G. (2013). “A Mechanism-Based Classification of Pain in Multiple Sclerosis,” Journal of Neurology, 260 (2) 351-67
Publisher – Google Scholar
Van’t Veer, A., Du, Y., Fischer, T. Z., Boetig, D. R., Wood, M. R. & Dreyfus, C. F. (2009). “Brain-Derived Neurotrophic Factor Effects on Oligodendrocyte Progenitors of the Basal Forebrain are Mediated Through TrkB and the MAP Kinase Pathway,” Journal of Neuroscience Research, 87 (1) 69-78
Publisher – Google Scholar
Vondran, M. W., Clinton-Luke, P., Honeywell, J. Z. & Dreyfus, C. F. (2010). “BDNF+/- Mice Exhibit Deficits in Oligodendrocyte Lineage Cells of the Basal Forebrain,” Glia, 58 (7) 848-856
Publisher – Google Scholar
VonDran, M. W., Singh, H., Honeywell, J. Z. & Dreyfus, C. F. (2011). “Levels of BDNF Impact Oligodendrocyte Lineage Cells Following a Cuprizone Lesion,” The Journal of Neuroscience, 31 (40) 14182-90
Publisher – Google Scholar
Vora, P., Mina, R., Namaka, M. & Frost, E. E. (2010). “A Novel Transcriptional Regulator of Myelin Gene Expression: Implications for Neurodevelopmental Disorders,” Neuroreport, 21 (14) 917-21
Publisher – Google Scholar
Willis, W. D., Al-Chaer, E. D., Quast, M. J. & Westlund, K. N. (1999). “A Visceral Pain Pathway in the Dorsal Column of the Spinal Cord,” Proceedings of the National Academy of Sciences of the United States of America, 96 (14) 7675-9
Publisher – Google Scholar
Zhang, X., Xu, Y., Wang, J., Zhou, Q., Pu, S., Jiang, W. & Du, D. (2012). “The Effect of Intrathecal Administration of Glial Activation Inhibitors on Dorsal Horn BDNF Overexpression and Hind Paw Mechanical Allodynia in Spinal Nerve Ligated Rats,” Journal of Neural Transmission, 119 (3) 329-36
Publisher – Google Scholar
Zhou, L.- J., Yang, T., Wei, X., Liu, Y., Xin, W.- J., Chen, Y., Pang, R.- P., Zang, Y., Li, Y.- Y. & Liu, X.- G. (2011). “Brain-Derived Neurotrophic Factor Contributes to Spinal Long-Term Potentiation and Mechanical Hypersensitivity by Activation of Spinal Microglia in Rat,” Brain, Behavior, and Immunity, 25 (2) 322-34
Publisher – Google Scholar
Zhu, W., Acosta, C., Macneil, B., Cortes, C., Intrater, H., Gong, Y. & Namaka, M. P. (2013). “Elevated Expression of Fractalkine (CX3CL1) and Fractalkine Receptor (CX3CR1) in the Dorsal Root Ganglia (DRG) and Spinal Cord (SC) in Experimental Autoimmune Encephalomyelitis (EAE): Implications in Multiple Sclerosis (MS) — Induced Neuropathic Pain (NPP),” In Press, Functional Neurology, 1-14
Publisher – Google Scholar
Zhu, W., Frost, E. E., Begum, F., Vora, P., Au, K., Gong, Y., Macneil, B., Pillai, P. & Namaka, M. (2012). “The Role of Dorsal Root Ganglia Activation and Brain-Derived Neurotrophic Factor in Multiple Sclerosis,” Journal of Cellular and Molecular Medicine, 16 (8) 1856-65
Publisher – Google Scholar