Introduction
Zinsser- Cole-Engman syndrome, also known as dyskeratosis congenita, is a rare genetic disorder characterized by triad of pigmentation and atrophy of the skin, leukokeratoses of oral mucosa and nail dystrophy along with bone marrow failure and predisposition to cancer [1]. The condition can be inherited as autosomal recessive, autosomal dominant and rarely X linked. It is proposed that mutations in dyskerin gene (DKC1) and telomerase RNA component (TERC) result in chromosomal instability and early cellular senescence with a progression to cancer [2]. Dermatologists should be able to recognize the early mucocutaneous features to arrive at the diagnosis, then, aiding frequent follow up to detect the malignancies at the earliest.
Case Report
Case 1
A ten year old boy, born out of non consanguineous marriage, presented to our department with history of asymptomatic skin lesions of 3 years duration. The skin lesions were pigmented and small to begin over the neck and chest, which gradually increased to involve other body parts. There were no similar complaints in the family. On examination, there were numerous hypo and hyperpigmented atrophic macules with mottled appearance over neck, limbs, and trunk. A few finger and toe nails were dystrophic. Palms and soles were smooth with numerous atrophic hypopigmented macules and absent dermatoglyphics. A few discrete white keratotic patches were evident over tongue. His systemic examination and routine hematological investigations were within normal limits.
Case 2
A 30 year old male, admitted under orthopedics for chronic hip pain, was referred to the dermatology department for his skin lesions. Skin lesions began appearing since the age of 10. These skin lesions were initially small and gradually progressed to attain the present size. On examination, numerous discrete to coalesced hypopigmented dry atrophic macules with mottled appearance were noted all over the body. Longitudinal ridges, pterygium, dystrophy, alternating red and white bands with terminal nicking of few finger and toe nails were seen. The skin of palms and soles was smooth with no dermatoglyphics. Discrete white patches were noted over tongue and buccal mucosa. Other mucosae were normal. X-ray of both hips showed evidence of degenerative changes of the head of the femur. Other systemic examination was normal. Routine hematological investigations were within normal limits.
Discussion
Zinsser- Cole-Engman syndrome, also known as dyskeratosis congenita, is a rare genetic disorder characterized by triad of pigmentation and atrophy of the skin, leukokeratoses of oral mucosa and nail dystrophy along with bone marrow failure and predisposition to cancer.[1] The condition can be inherited as autosomal recessive, autosomal dominant and is rarely X linked. The exact pathomechanism of this condition is not fully understood. It is proposed that mutations in dyskerin gene (DKC1) and telomerase RNA component (TERC) result in poor telomere maintenance which is central to the pathogenesis of this condition. Dyskerin gene is located on X chromosome at Xq28. Low levels of TERC in patients of dyskeratosis congenita lead to chromosomal instability and early cellular senescence with a progression to cancer.[2] Rapidly proliferating cells, which need high telomere maintenance like bone marrow cells,
The histology of pigmented skin lesions usually show epidermal atrophy, dilated capillaries and pigment laden macrophages in the upper dermis. The leukokeratotic lesions of the tongue may show epithelial dysplasia in early age, and carcinoma in situ to frank malignancy in late stages of the disease. [3,4]
This condition is more common in males than females with a male to female ratio of 3:1. The mucocutaneous findings are the first ones that develop as early as 5years. The predominant cutaneous finding is hypopigmented or hyperpigmented atrophic macules and patches with telangiectasia in a mottled or reticular pattern over sun-exposed and unexposed areas. This reticular pigmentation is more pronounced over the neck giving rise to dirty neck appearance.[4] The palms and soles may be thickened and hyperhidrotic with absence of dermatoglyphics and may form bullae even with minimum trauma. The minor cutaneous findings include cicatricial alopecia and premature canities. The teeth tend to be defective, irregularly implanted with periodontal disease and early caries. Nail dystrophy is seen in the majority of cases with finger nails getting affected earlier than toe nails. This is often preceded by ridging and longitudinal splitting. Pterygium and anonychia are the end-stage changes. [1]
Mucosal lesions also appear early in the course of the disease which coincide and or follow nail and skin changes. It is characterized by small blisters and ulcerations leading to leukokeratosis over the tongue, buccal, oropharyngeal and genital mucosa. Leukokeratosis of oropharynx, oesophagus leads to dysphagia. Similar changes on the tarsal conjunctiva may obliterate the lacrimal puncta, resulting in epiphora, soreness and scarring of the lids. One should look for development of malignancies in these patients especially squamous cell carcinoma which usually arises out of long standing lesions of leukokeratosis of the mucosa.[1,5]
Avascular necrosis of hip bones, as seen in our second case, is one of the major skeletal manifestations of this condition requiring surgical correction.[6] Other skeletal changes are hypoplasia of mandible and scoliosis. Gastrointestinal findings include: esophageal webs, hepatosplenomegaly, and cirrhosis. [7] Genitourinary manifestations include meatal stenosis leading to acute urinary obstruction, testicular atrophy and hypospadias.[8] Female carriers of dyskeratosis congenital usually have subtle findings like, single nail dystrophy, solitary hypopigmented patch or mild leukokeratosis. The severe variant of X-linked dyskeratosis congenita (Hoyeraal- Hreidarsson) syndrome is characterized with cerebellar hypoplasia and can cause enterocolitis in infancy.[9] Dermatologists should be able to pick up the findings in the earlier stages, anticipate the complications and refer the patients to subsequent specialties.
This condition has to be differentiated from other causes of poikiloderma, such as Weary- Kindler syndrome and Rothmond-Thomson syndrome. In the former, the skin has a parchment-like appearance, and the later has poikiloderma over the buttocks from a very young age and micky mouse ear.[1,]
There is no effective treatment for this condition. Methods aiming at increasing TERC expression can be promising. [10] The prognosis is bad in these patients as it is a multisystem disorder and death occurs mainly due to bone marrow failure, infection and malignancy. [1,4]
Conclusion
These cases have been reported for their rarity. The authors would like to emphasize the need for detecting early signs of the condition, to advise regular follow up and to tackle complications that may develop.
Figures
Fig 1a & 1b: Reticular network of atrophic pigmented lesions over neck giving dirty neck appearance
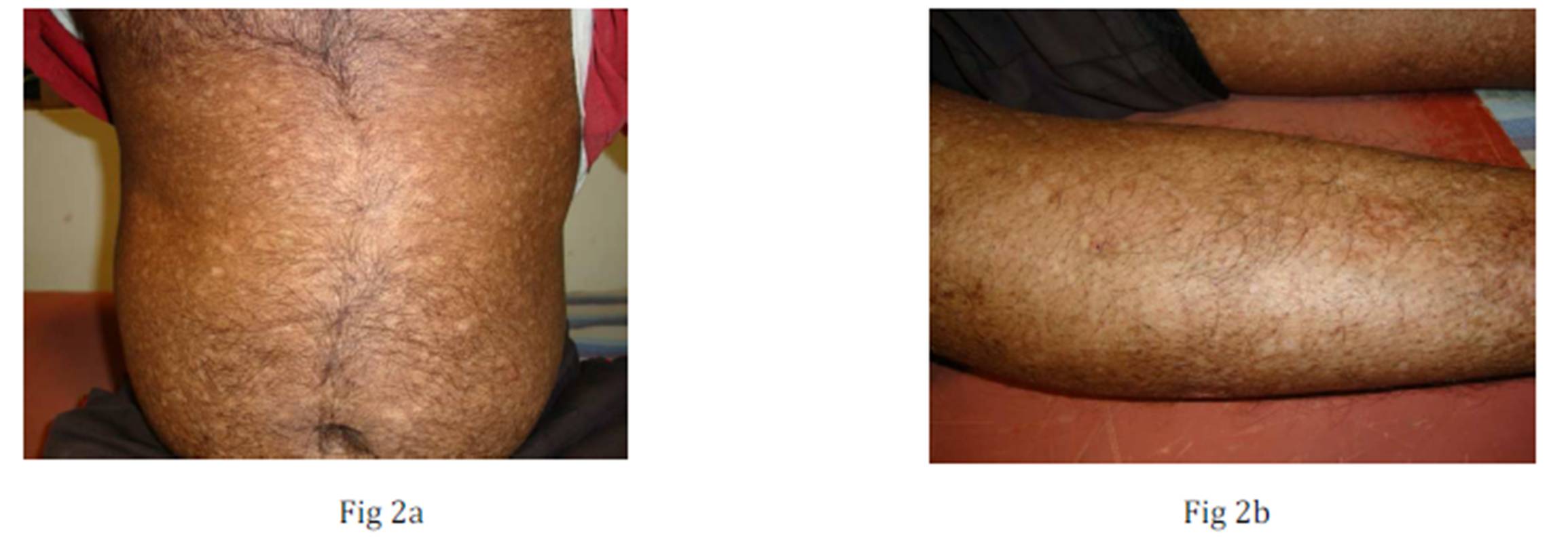
Fig 2 a & 2b: Multiple discrete atrophic macules over trunk and limbs
Fig 3 a & 3b: Multiple leukokeratotic lesions over tongue
Fig 4 a & 4b: Ridging and pterygium of finger nails and dystrophy of toe nails
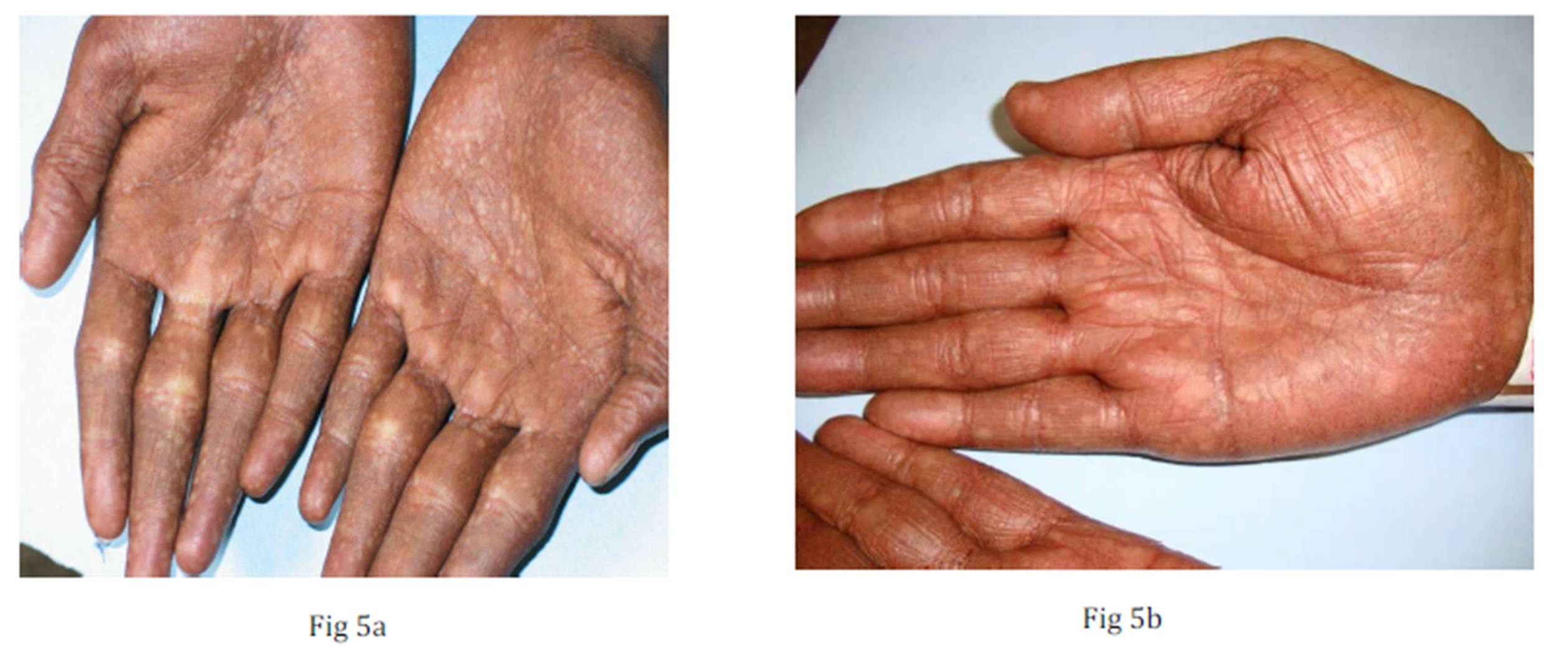
Fig 5 a & 5b: Atrophic macules over palms with absent dermatoglyphics
Reference
1. Irvine AD. Mellerio JE (2010) Genetics and Genodermatosis. In: Rook’s text book dermatology. Burns T, Breathnach S, Cox N, Griffiths C edrs. 8th edn, Wiley- Blackwell 15.1-15.97.
2. Walne AJ, Marrone A, Dokal I (2005). Dyskeratosis congenita: a disorder of defective telomere maintenance? Int J Hematol 82:184-9.
Publisher – Google Scholar
3. Joshi RK, Atukorala DN, Abanmi A et al (1994). Dyskeratosis congenita in a female. Br J Dermatol130:520-22.
Publisher – Google Scholar
4. Moss C (2011) Rothmond-Thomson syndrome, Bloom syndrome, Dyskeratosis congenita, Fanconi anaemia. In: Harper’s text book of pediatric dermatology. Irvine A, Hoeger PH, Yan AC edrs, 3rd edn, Wiley- Blackwell; 136.1-136.13.
5. Sawanth P, Chpada NM, Desai DC et al (1994). Dyskeratosis congenital with esophageal stricture and dermatological manifestations. Endoscopy 26:711-12.
Publisher – Google Scholar
6. Klab RE, Grossman ME, Hutt C (1986) Avascular necrosis of bone in dyskeratosis congenita. Am J Med; 80(3):511-3.
Publisher – Google Scholar
7. Redkar NN, Pandey DB, Jerajani HR, Padhiyar R, Dhokare A. Dyskeratosis congenita with portal hypertension of unknown etiology. J Assoc Physicians India. 2011; 59:260-3.
8. Oslen TG, Peck GL, Lovogrove RH (1981). Acute urinary tract obstruction in dyskeratosis congenita. J Am Acad Dermatol; 4:556-60.
Publisher – Google Scholar
9. Borggraefe I, Koletzko S, Arenz T et al (2009). Severe variant of X-linked dyskeratosis congenital (Hoyerral-Hreidarsson syndrome) causes significant enterocolitis in early infancy. J Pediatr Gastroenterol Nutr; 49:359-63.
Publisher – Google Scholar
10. Agarwal S, Loh YH, McLoughlin EMet al (2010), Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature; 464: 292-296.
Publisher – Google Scholar