Introduction
Ciliopathies are a group of overlapping disorders whose etiologies lie in defective structure and function of cilia (Davis and Katsanis, 2012). The cilia are organelles found on the apical surface of most eukaryotic cells playing essential roles during development and tissue homeostasis (Garcia-Gonzalo and Reiter, 2012). The ubiquitous presence of cilia in every cell of body tissues explains the wide range of human diseases arising from defects in cilia structure or function. Classically, the function of cilia has been attributed to motility, with propelling cell locomotion or generating flow of fluid environment (Malicki, 2012). Recently, cilia have found identified as important sensory organelles involved in signaling pathways (Yuan and Sun, 2013). Furthermore, recent discoveries have assigned novel functions to primary (nonmotile) cilia, ranging from participation in different signal transduction pathways through extracellular receptors and maintaining cellular homeostasis and cell division which direct embryonic development and organ function (Kim and Dynlacht, 2013). Because cilia, either primary or motile cilia, are present in nearly all cell types of all organs, the defects in these organelles lead to numerous and uncommon human diseases grouped collectively as ciliopathies. The production of specific disorders by ciliopathies is nowadays under research. We can speculate that mutations in ciliary gene can lead to defects in various signaling pathways. Recently, defects in proteasomal clearance of specific proteins have linked to several ciliary phenotypes (Liu et al, 2014). Thus, it is a rapidly expanding field of research involving multiple disciplines, which will improve diagnostic testing and identify new therapeutic targets. Understanding of the genetics, molecular abnormalities, and clinical manifestations in children will improve the diagnostic awareness and consequently the evidence based treatment. Our aim was integrate both clinical and molecular aspects of these genetic diseases highlighting the novel genotype-phenotype associations to facilitate their recognition by pediatricians. This review summarizes the recent advances in these interesting conditions which may explain the clinical manifestations and the understanding of these processes in childhood.
Structure and Function of the Cilium
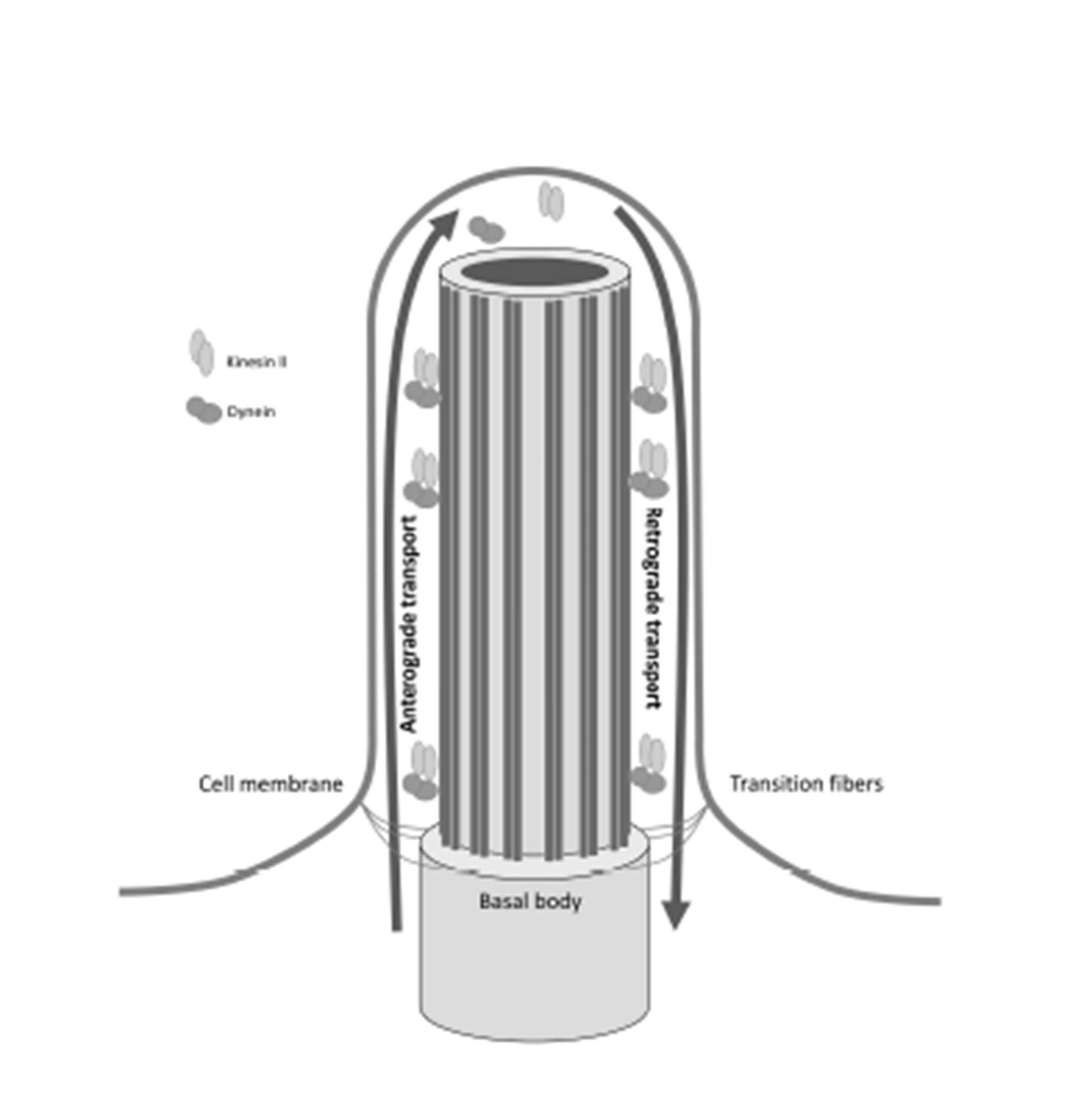
Cilia are organelles conserved throughout evolution and are present in most cells of the human body. Emerging from the basal body, a centriole derived structure; the cilium extends from the cell surface into the extracellular space and is composed of a microtubular proteic structure known as the axoneme. The basal body contains 9 pairs of peripheral microtubule cilia anchored to the cell surface, and the orientation of these cilia determines ciliogenesis (Figure 1) (Bandano et al, 2006; Waters and Beales, 2010; Hildebrandt et al, 2011; Ferkol and Leigh, 2012).
Figure 1: Microtubular structure of the cilium and intraflagellar transport. The ciliary axoneme is a microtubule structure anchored to the basal body and surrounded by the ciliary membrane. Molecular complexes kinesin II and cytoplasmic dynein are involved in intraflagellar transport and their function is required for proper ciliogenesis.
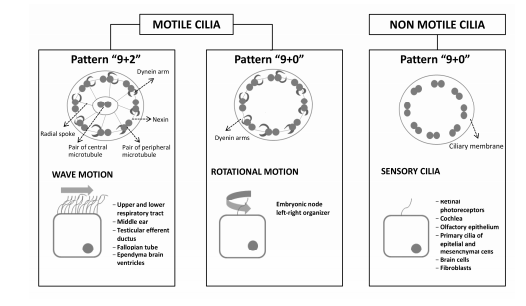
Cilia are classified into three principal types based on microtubule structure: motile cilia with a “9+2” axoneme pattern, motile cilia with a “9+0” axoneme pattern, and immotile cilia, also called sensory or primary cilia, with a “9+0” axoneme pattern. Recent studies have shown that both motile and immotile cilia can carry out sensory functions in the organism; we use the term “sensory cilia” to refer to non motile monocilium or primary cilium (Hildebrant et al, 2011; Fliegauf et al, 2007).
Cilia are organized in a microtubular structure, formed by helical protofilaments made up of monomers of alpha and beta tubulin. Motile cilia exhibit a whipping motion and are involved in respiratory tract mucociliary clearance, cerebrospinal flow movement and transport of ovum and sperm in the reproductive tracts. These cilia have the classical “9+2” structure: 9 pairs of peripheral microtubule surrounding a central pair all contained in the cell membrane. Each peripheral doublet microtubule contains one external and one internal dynein arm. Dynein arms contain ATPase, which drives the sliding movements between the peripheral microtubule doublets. Nexin joints limit movement in adjacent ciliary doublets maintaining intact cilium during motion. Central tubules, wrapped in a central sheath, are joined by peripheral doublet radial arms, controlling the activity of dynein arms and maintaining the structure of the cilium (Figure 2) (Fliegauf et al, 2007; Water and Beales, 2010; Hildebrandt et al, 2011).
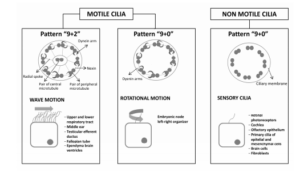
Figure 2: Motile cilia and non motile cilia: ciliar pattern, type of movement and system location. Motile “9+2” pattern cilia are located on the cell surface as multiple cilia and have a synchronized waveform motion. Motile “9+0” pattern cilia lack central microtubules and are responsible for establishing the left-right axis in the embryo. Immotile cilia “9+0” lack dynein arms and central microtubules and act as primary or sensory cilia in multiple organs of the body.
Motile cilia with a “9+2” pattern have a synchronous motion waveform, whose direction depends on the orientation of the pair of central microtubules. The low viscosity of the periciliary liquid found on the epithelial surface allows for rapid beat frequency. The ciliary beat is coordinated by calcium signals between epithelial cells through gap junctions.
Motile cilia with a “9+0” pattern lack central microtubules and are found only in the ventral node during embryonic gastrulation. These cilia have a rotational movement and are responsible for generating the extracellular leftward flow, which establishes the left-right axis in the embryo.
Sensory primary cilia follow a pattern “9+0” pattern and lack the central microtubules and dynein arms. They function as single cilium on the surface of most vertebrate cells. Primary cilia have extracellular receptors, which can act as chemo-or mechanoreceptors and operate in response to light, temperature or gravity stimuli. They are also involved in numerous signaling pathways responsible for the development and homeostasis of various tissues (Bisgrove and Yost, 2006; Berbari et al, 2009).
There are other patterns in the ciliary axoneme, as sensory cilia “9+2” present in the vestibular system and a nobel “9+4” axoneme pattern indentified by ultrastructural microscopical analysis on the notochordal of rabbit embryo. It shows that even though the cilia are evolutionarily conserved organelles, the structure of the axoneme may vary among different species of vertebrates (Feistel and Blum, 2006).
Intraflagellar Transport (IFT)
Synthesis of the structural and functional elements of the cilium takes place in the cytoplasm. Assembly of protein elements is accomplished by intraflagellar transport along the ciliary axoneme. The IFT particles are composed of two complexes; IFTA with 6 protein subunits is needed for retrograde transport and IFT B with 13 protein subunits which is responsible for anterograde transport. Protein components are assembled in the basal body and undergo anterograde transport to the upper end of the cilium driven by the kinesin II motor complex. At the upper end of ciliary axoneme, the kinesin II motor complex is inactivated, facilitating retrograde transport to the base of the cilium through cytoplasmic dynein. Disruption of IFT complex or basal body proteins leads to abnormal cilia assembly and function disorders (Berbari et al, 2009; Bisgrove and Yost, 2006) (Figure1).
Ciliopathies
Mutations in over 100 ciliary genes has been identified as a causative of several groups of diseases due to the disfunction of cilia, collectively known as ‘‘ciliopathies,’’ that often share common phenotypic features. As a multitude of genes are required for the construction of cilia and the centrioles from which cilia originate, ciliopathies serve as a model for the complex genetic interactions seen in human genetic diseases (Pan et al, 2005; Sharma et al, 2008; Berbari et al, 2009).
Motor Ciliopathies
The motor ciliopathies include alterations in the embryonic formation of the left-right axis, primary ciliary dyskinesia, and dysregulation in cell division and oncogenesis (Table 1).
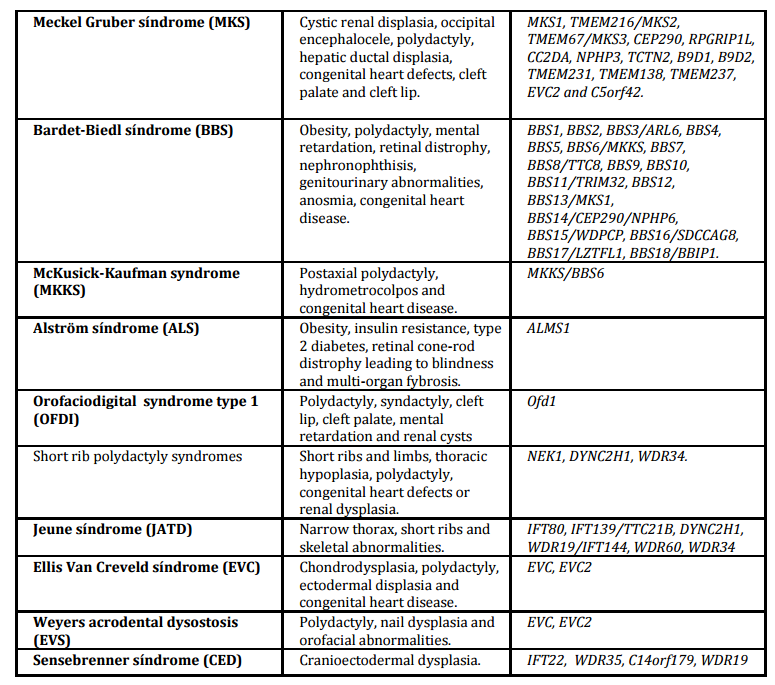
Table 1. Ciliopathies: clinical manifestations, syndromes and genes involved
Left-Right Axis Formation in the Embryo
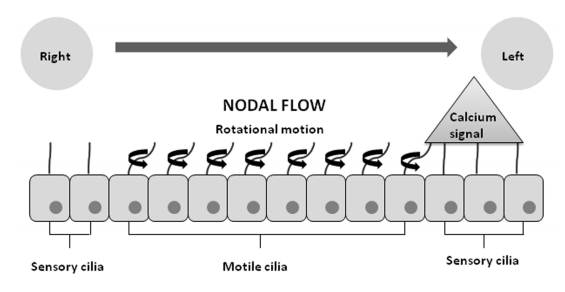
Embryonic gastrulation proceeds as a symmetrical process until the initial break at Hensen’s node. On the ventral surface of the node, there are two types of cilia: motile “9+0” pattern cilia and immotile “9+0” pattern cilia. The motile cilia are located in the central node and move rotationally to produce a leftward flow of perinodal fluid. This flow is responsible for the onset of the signaling pathway leading to leftwards orientation formation. As axonema move in response to the perinodal flow that follows a right-left direction, a cascade of intracellular calcium signaling on the left side of the node is triggered; immotile cilia found in the periphery of the node act as mechanoreceptors in response to this signal. The intracellular calcium signal requires the presence of the ciliary protein polycystin-2 (PKD2) (Figure 3), which is thought to be involved in mechanosensation. In this way, the nodal flow might contribute to left-right asymmetry (Afzelius, 1995; Icardo et al, 2002).
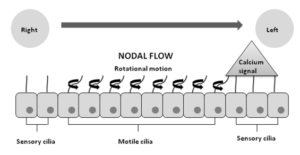
Figure 3: Left-right axis establishment in the embryo, Cilia flexion triggers a left-sided intracellular calcium signal
Primary Ciliary Dyskinesia
Primary ciliary dyskinesia (PCD, MIM ID#244400), or immotile ciliary syndrome, was the first clinical entity known and associated with ciliary dysfunction. It includes a group of diseases in which dyskinetic and ineffective ciliary motion, or even ciliary aplasia, render respiratory cilia are immotile (Zariwala et al, 2007).
The cilium cells present in the nasopharynx, paranasal sinuses, middle ear and respiratory tract from the trachea to the bronchioles. Each single cilium cell has approximately 200 cilia on its surface that beat in a coordinated manner to achieve mucociliary clearance. Dysfunction of respiratory ciliated cells leads to chronic respiratory tract infections and accumulation of mucous secretions present in the majority of cases since birth. As the disorder also affects the ciliary motility of the sperm flagellum and the motility of cilia in the fallopian tubes, male sterility and reduced fertility in women is rather common among the affected. Because of the inefficiency of the nodal cilia in embryonic establishment of the left-right axis, approximately 50% of these patients have a total situs inversus or situs ambiguous (called Kartagener´s syndrome, MIM ID#244400), (Afzelius, 1995; Kennedy et al, 2007).
Primary ciliary dyskinesia is considered an autosomal recessive disorder, although rare cases have been described with autosomal dominant or X-linked transmission, the latter in relation to the gene RPGR (retinitis pigmentosa GTPase regulator) and OFD1 gene which has been found in a family with respiratory ciliary dyskinesia, macrocephaly and mental retardation (Coene et al, 2009). Its incidence is estimated at 1 in 15,000-30,000 live births. Theoretically, any mutation in the hundreds of protein elements that constitute the complex ciliary structure could cause PCD. The most common ultrastructural defect is the complete or partial absence of dynein arms; approximately two thirds of patients have a defect in outer dynein arm. Other less common causes of PCD include defects of radial joints in the arms of nexin, transposition of ciliary microtubules or agenesis. It should be noted that normal ultrastructure is observed in 15% of PCD cases associated with DNAH11 mutations.
PCD is a genetically heterogenous disorder and over 28 genes have been identified (table 1) DNAI1 and DNAH5 mutations are responsible of 25% PCD pacients and almost 50% with defects in outer dynein arm, making these genes a target of genetic screening (Popatia et al, 2014; Cardenas-Rodriguez and Badano, 2009; Leigh et al, 2009).
A prenatal finding of total situs inversus finding could be an indication of PCD. Neonatal respiratory distress and constant rhinorrhea could be the first clinical manifestations of this disease. Common childhood manifestations are primary chronic productive cough with daily mucopurulent sputum production, chronic sinusitis and secretory middle ear infection, leading to varying degrees of conductive hearing loss. Nasal polyposis may be present. Defects in mucociliary clearance lead to recurrent infections of the lower respiratory tract with recurrent pneumonia caused by Haemophilus Influenzae, Staphylococcus Aureus and Streptococcus Pneumoniae. Pseudomonas Aeruginosa infection is more common in older patients and in advanced stages of the disease. The finding of “idiopathic” bronchiectasis, found in only 10% of preschool patients, is an important clinical data that lead to the diagnosis of PCD. Despite the prominent clinical manifestations, the average age of diagnosis in most studies is 4 years. Commonly, these patients also suffer from recurrent headaches secondary to chronic sinusitis. However, in very rare cases the headaches are caused by hydrocephalus secondary to cerebral ependyma, which is also associated with cilia dysfunction (Davenport and Yoder, 2008; Baker and Beales, 2009).
Cilia and Cell Division: Role in Oncogenesis
Several tumor phenotypes can be adjusted to ciliary dysfunction spectrum, in which case the ciliar loss would be a necessary prerequisite for cell proliferation. However, depletion of cilia formation alone is not a sufficient event to drive tumorigenesis. The cilium is involved in multiple signaling pathways allowing coordination of different cell types. Cilia dysregulation of cells is important to oncogenesis because of the role that centrosome amplification and subsequent genomic instability play in many cancers (Basten and Giles, 2013).
The current view suggests that primary cilium is a sensor for the luminal fluid of the nephron. This mechanosensation is important in maintaining tubular architecture because extracellular signals are transformed into specific signaling pathways that regulate tubular cell polarization and differentiation. There are multiple evidences suggesting that the presence of renal cysts would be a precursor stage of tumor formation. Kidney cysts are present in several classic ciliopathies as autosomal dominant polycystic kidney disease, Bardet-Biedl syndrome, nephronophthisis and orofaciodigital type 1 syndrome that are described below. Von Hippel-Lindau disease (VHL, MIM ID#193300) is an autosomal dominant condition that is caused by inactivation of the VHL gene and is implicated in most sporadic clear cell renal carcinomas. Mutations of the VHL gene on the primary cilium lead to kidney cysts and predispose to diverse tumors, suggesting that VHL acts as a tumor suppressor (Egeberg, 2012). Tuberous sclerosis (TSC1, MIM ID#191100; TSC2, MIM ID#613254) is a tumor suppressor gene syndrome associated with renal cyst formation and tumorigenesis in various organs including the kidney, brain, retina and skin. TSC is associated with germline mutations in TSC1/Hamartin and TSC2/Tuberin. The renal manifestations in TSC include benign renal angiomyolipoma (50 to 80%) and a minority of subjects develops clear cell renal carcinoma (Egeberg, 2012). The monogenic disorder Birt-Hogg Dubé (BHD, MIM ID#135150) is an autosomal dominant characterized by the development of renal cysts, kidney cancer, pulmonary cysts and bening skin tumors (fibrofolliculomas). BHD disease is caused by heterozygous mutations in the BHD gene encoding folliculin (FLCN) a protein considered a tumor suppressor. In contrast to TSC1/2, FLCN does not affect cilia length. Alteration of FLCN levels can cause changes to the onset of ciliogenesis by alteration of “canonical Wnt signaling” (Esteban et al, 2006; Furuya and Nakatani, 2013).
In breast cancer has been shown that ciliary frequencies are decreased when compared to normal breast tissue and cilia, associated genes Gli1 (Hh effector), RPGRIP1 (LCA) and DNAH9 (PCD) are commonly mutated in this type of cancer. In the development of melanoma in situ and pancreatic ductal adenocarcinoma has been observed ciliary progressive loss at different stages of tumor (Basten and Giles, 2013). Colorectal cancer has been associated with ciliary dysfunction. The protein kinase Aurora A involved in cilia loss is frequently mutated in this cancer (Furuya and Nakatani, 2013). Murine studies demonstrate the involvement of the cilia in the development of skin tumors of the basal cell carcinoma subtype and medulloblastoma brain tumors by the “Hh signaling pathway” (Toftgård, 2009; Wong et al, 2009).
Sensory Ciliopathies
Sensory primary cilia are involved in multiple biological processes. They act as chemoreceptors or extracellular mechanoreceptors and are involved in the process of cell division, serving as a structural element for the formation of the mitotic spindle and regulating the process of cell proliferation and apoptosis (Marshall and Nonaka, 2006; Singla and Riter, 2006; Quilan et al, 2008). Furthermore, it has been demonstrated the involvement of primary cilium in several signaling pathways, including hedgehog (Hh), canonical Wnt and planar cell polarity, platelet-derived growth factor, fibroblast growth factor, Notch and Hippo cascades (Ashe et al, 2012).
There are several syndromes, many with overlapping clinical features that are associated with primary cilium dysfunction. The main sensory ciliopathies, genes involved and clinical features of each syndrome are presented in Table 1 (Eggenschwiler and Anderson, 2007; Gerdes et al, 2009; Ferkol and Leigh, 2012).
Renal and Liver Ciliopathies: Polycystic Kidney Disease and Congenital Hepatic Fibrosis
The autosomal dominant polycystic kidney disease (ADPKD, MIM ID#173900) was one of the first clinical entities associated with primary cilia dysfunction. ADPKD affects 1 in 500-1000 live births. The disorder is characterized by the progressive growth and development of renal cysts that destroy functional parenchyma, leading to dilation of renal collecting tubules and kidney failure. Cysts development begins in utero and progresses slowly, is often no evident until adult age. Mutations in two genes, PKD1 and PKD2, have been described in association with this entity. The PKD1 gene, which encodes the protein polycystin 1, is the most frequent alteration found in up to 85% of all cases and specifically associated with severe cases of the disease. The gene encoding the protein PKD2 polycystin 2 is also involved in establishing the left-right axis during embryonic development (Waters and Beales, 2010; Hildebrandt et al, 2011).
The primary cilium was first suspected in cystic disease after observations that most proteins implicated in the pathogenesis of the disease which are part of the molecular structure of the cilium and are involved in cystogenesis. The primary cilium can act as a flow sensor in the renal tubule. Ciliary deflection occurs in response to renal flow, triggering the entry of calcium into the cell, a process mediated by polycystin 2 (PC2), which acts as a cation channel with polycystin 1 (PC1). Intraflagellar transport is also required to regulate ciliogenesis and the levels of PC2 at the cilium. Furthermore, the absence of renal flow induces COOOH-terminal proteolysis of PC1. In this way, renal cilia are essential for PC1’s function as a mechanoreceptor and may modify gene transcription of PC1. The low levels of intracellular calcium lead to an increased expression of PC1 and subsequent migration of aquaporin-2 to the apical membrane increasing osmotic permeability. These data show the role of primary cilium as a mechanosesnor in the lumen of the collecting duct tubule (Eccles and Stayner, 2014; Pan et al 2005; Waters and Beales, 2010).
Autosomal recessive polycystic kidney disease (ARPKD; MIM ID#263200) is the most common ciliopathy in children. It is characterized by dilation of the collecting ducts of the kidney, renal cystic degeneration and progressive liver fibrosis. Most patients die within the first year of life. Liver disease is an invariable manifestation of ARPKD syndrome and can be a predominant clinical feature (Eccles and Stayner, 2014; Torres and Harris, 2009).The disease is caused by mutations in the gene PKHD1 encoding the protein polyductin or fibrocystin, which is responsible for the differentiation of cells lining the renal collecting ducts. Abnormalities in defective cilia-based signaling are common to renal and biliary epithelia and may be present in different ciliopathies that are related below (Gunay-Aygun, 2009).
Cholangiocytes, the epithelial cells lining intrahepatic bile ducts, have primary cilia detecting changes in bile flow and osmolality. Cholangiocyte cilia are sensory organelles responding to mechanical stimuli by alterations in intracellular Ca2+ signal and cAMP. These cilia can also detect changes in composition and tonicity of bile and play an important role in ductal bile formation by acting as osmosensors. The osmosensory function of primary cilia in biliary epithelia is associated with the function of transient receptor potential vanilloid 4 (TRPV4) Ca2+ ciliary channel, and is also linked to ATP release. Bile tonicity detected by the osmosensor proteinTRPV4 expressed on cholangiocyte is the main mechanism for bicarbonate secretion in bile ducts and determine the bile duct formation (Gradilona et al, 2007; Masyuk et al, 2008).
Congenital hepatic fibrosis/Caroli’s syndrome (CHF/CS) is an autosomal recessive malformation characterized by hepatic fibrosis, portal hypertension and renal cysts. Pathologically is defined by periportal fibrosis and irregularly shaped proliferating bile ducts. In most cases, CHF/CS presents as part of a multisystem disorder usually associated with ciliopathies that have associated renal disease: ADPKD, nephronoptisis, and chronic tubulointerstitial disease, collectively referred to as the hepatorenal fibrocystic diseases and rarely appears as an isolated clinical manifestation (Gunay-Aygun, 2009; Shorbagi and Bayraktar, 2010).
Nephronophthisis and other Associated Ciliopathies
Nephronophthisis (NPHP) is an autosomal recessive tubulointerstitial nephropathy being the most common genetic cause of chronic kidney disease in the first three decades of life. Initially, patients affected usually present symptoms of polyuria and polydipsia with secondary enuresis and anemia. NP can be classified clinically in infantile, juvenile and adolescent by the onset of end-stage renal failure. The presentation occurs typically during early puberty with progressive renal failure. Kidneys in NPHP have normal sized with loss of cortico-medullary differentiation. Histologically it has been identified the presence of cortico-medullary cysts, tubulointerstitial cell infiltrates and tubular basement membrane disruption.
Mutations have been reported in 12 genes (NPHP1-11 and NPHPL1), which account approximately 30% of cases of NPHP. These genes encode nephrocystin proteins that are located in the cilium, basal bodies and centrosome or adherens junctions between cilium cells (Arts and Knoers, 2013). NPHP1 mutations are the most common cause of NPHP which is responsible of NPHP juvenile type and is reported in about 20% of all patients affected. NPHP1 encodes nephrocystin-1 which is located at adherens junctions of renal epithelial cells (Hildebrant et al, 2009).
NPHP2 causes infantile NPHP and can be associated with situs inversus (Waters et al, 2011). NPHP2 encodes inversin protein which is necessary for planar cell polarity and normal tubular development of renal tubular cells. It has been demonstrated the interaction between nephrocystin-1, inversin and β-tubulin in the microtubule axoneme of the primary cilia explaining the pathogenesis of NPHP, left-right axial determination and primary cilia function (Hildebrant et al 2009, 2011).
NPHP3 is responsible of adolescent NPHP. Truncation of NPHP3 function results in severe multisystem disorder with embryonic lethality in mice associated with situs inversus and heart defects (Bergmann et al, 2008)
NPHP4 encodes nephrocystin- 4 protein and has been found in juvenile NPHP. NPHP4 can interact with other ciliopathy proteins, as RPGRIP in Leber congenital amaurosis and RPGRIP1L in Joubert syndrome (Hildebrant et al 2010). NPHP5 encodes protein nephrocystin-5 which interacts with calmodulin and RPGR (retinitis pigmentosa GTPase regulator) providing the association with X-linked retinitis pigmentosa disorders (Otto et al, 2005).
NPHP 6/CEP290 encodes a protein located in centrosoma and mitotic spindles. NPHP type 6 may be associated with retinal degeneration and cerebelar defects with or without renal involvement (Hildebrant et al 2011).
NPHP7/GLIS2 is envolved in Hedgehog signaling pathway by the protein Glis2 that controls cell determination during embryogenesis. In the absence of the protein Gli2, a key signaling component of this pathway, renal epithelial dysregulation leads to fibrosis (Hidebrandt et al, 2011; Wolf and Hildebrandt, 2011).
NPHP patients often have extrarenal manifestations including retinitis pigmentosa, congenital hepatic fibrosis, left-right laterality defects, cerebellar vermis hypoplasia and skeletal abnormalities as we see below (Hurd and Hildebrant, 2011).
Joubert Syndrome
Joubert syndrome (JBTS, MIM ID#213300) is a rare syndrome characterized by congenital malformation of the brain stem, cerebellar vermis agenesis or hypoplasia causing hypotonia, ataxia psychomotor delay, irregular breathing patterns and oculomotor apraxia. Additional clinical features include NPHP, retinal degeneration, ocular colobomas, polydactyly or endocrine abnormalities. The defining characteristic of Joubert syndrome is the “molar tooth sign” on cranial magnetic resonance imaging a consequence of hypoplasia of the cerebellar vermis and malformations of the midbrain and hindbrain. The variable clinical manifestations associated with the molar tooth sign don´t comprise different disorders; they are part of the wide clinical range of Joubert syndrome. Genetic complexity in Joubert syndrome reveals the phenotypic variability of this syndrome, and therefore using the term” Joubert syndrome and related disorders” only leads to confusion in diagnosis (Romani et al 2013)
The disease has an autosomal recessive inheritance pattern and an incidence estimated at 1 in 100,000 births. It is also a genetically heterogeneous syndrome; mutations have been identified in 23 genes all of which encode for proteins of primary cilium and are responsible of about half of cases (Table 1). Mutations in TMEM67 correlate in about 80% of patients with Joubert syndrome and hepatic involvement. Mutations in CC2D2A or RPGRIP1L are correlated with the disorder known by the acronym COACH (cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma and hepatic fibrosis). Finally, mutations in CEP290 are present in about 50% of patients with JS and cerebello-oculo-renal phenotype, including retinopathy and juvenile nephronophthisis (Sattar and Gleeson, 2011; Valente et al, 2013; Coene et al, 2009). Recently, two other mutations in MKS1 and B9D1 genes implicated previously with Meckel syndrome have been identified in patients with mild Joubert phenotype (Romani et al, 2014).
There is a genetic and clinical overlap with other ciliopathies, the most important is the relation with Meckel syndrome, a lethal ciliopathy characterized by cystic kidneys, bile duct proliferation, encephalocele and polydactyly sharing 13 genes causative of both syndromes. Other ciliopathies with clinical and genetic overlap are Senior-Loken syndrome, Bardet-Biedl syndrome, isolated nephronophthisis and some orofaciodigital and skeletal syndromes (Valente et al, 2013).
Senior-Loken Syndrome
Senior-Loken syndrome (SLS, MIM ID#266900) is a rare disease with autosomal recessive inheritance pattern characterized by the association of nephronophthisis and retinitis pigmentosa or different degrees of retinal distrophy. Rarely, other clinical signs may be observed further in life including liver fibrosis, obesity and neurological diseases.
SSL is also a genetically heterogeneous ciliopathy. Mutations have been described in several genes called nephrocystins (NPHP1, INVS/NPHP2, NPHP3, NPHP4, IQCB1/NPHP5, CEP290, NPHP6, GLIS2/NPHP7, RPGRIP1L/NPHP8, NEK8/NPHP9, SDCAAG8/NPHP10, TMEM67/NPHP11, TTC21B/NPHP12, WDR19/NPHP13). These genes encode proteins present in the primary cilia of kidney cells and in the connecting cilia of photoreceptor cells. NPHP1 gene deletions are the most common abnormality. The incidence of RP with nephronopthisis is dependent on the mutated gene and it reaches 100% of cases in NPHP5 and NPHP6 gene mutations (Ronquillo et al, 2012).
Leber Congenital Amaurosis
Leber congenital amaurosis (LCA, MIM ID#204000) is the most severe retinal dystrophy leading blindness in the first year of life. It has been identified mutations in 14 genes (table 1) as causative of LCA encoding proteins involved in different retinal pathways. The most frequently mutated are CEP290, GUCY2D and CRB1 (Den Hollander et al, 2008; Wang et al 2009). The retinal disease of LCA may present as a clinical feature in other syndromes like Joubert syndrome or Senior-Loken syndrome (Waters, 2011). Recently, it has opened a hope in those patients with experimental gene therapy in RPE65 in dogs restoring sight in this animals models (Koenekopp, 2004).
Meckel-Gruber Syndrome
Meckel-Gruber syndrome (MKS, MIM ID#249000) is one of the most severe ciliopathies with lethality in perinatal period. It is characterized by cystic renal disease, occipital encephalocele, polydactyly and hepatic fibrosis. The incidence is 0.62 in 100,000 births (Martinez-Frías et al, 2012).
MKS is a heterogeneous genetic syndrome; mutations in MKS1, TMEM216/MKS2, TMEM67/MKS3, CEP290, RPGRIP1L, CC2DA, NPHP3, TCTN2, B9D1, B9D2, TMEM231, TMEM138, TMEM237, EVC2 and C5orf42 have been identified in this syndrome. There is an important overlap in genes involved in JBTS, NPHP and MKS even though the clinical presentation of these syndromes is different (Baker et al, 2014; Romani et al, 2013; Parelkar et al, 2013; Dowdle et al, 2011; Tammachote et al, 2009)
Bardet-Biedl Syndrome
Bardet-Biedl syndrome (BBS; MIM ID#209900) is a ciliopathy with high variability of clinical features including obesity, retinal distrophy, postaxial polydactyly, renal anomalies, hypogonadism and mental retardation. Clinical manifestations may occur later during childhood hindering its early diagnostic. The principal cause of morbidity and mortality in BBS patients is renal failure due to cystic tubular disease and anatomical malformations. Secondary features include ataxia, anosmia, diabetes, cardiovascular anomalies or Hirschprung disease. The clinical spectrum of this syndrome is determined by its genetic heterogeneity. It has been described that mutations in 18 genes (BBS1 to BBS18) and in most cases are autosomal recessive inheritance (Table 1). These genes encode primary cilia proteins that have functions in the maintenance of microtubule structure and coordination of the cell cycle. The recent identification of mutations in MKS1 gene in BBS has supported the assumption that MKS can represent a severe BBS phenotype (M´hamdi et al, 2014; Beales et al, 1999; Ansley et al, 2003; Dawe et al, 2007).
It was recently demonstrated in adult mice that ciliary dysfunction in the protein IFT88 implicated in intraflagellar transport can induce hyperphagia and obesity interfering in the function of proopiomelanocortin neurons in the hypothalamus. Rahmouni et al (2008) showed that BBS mice have high levels of plasma leptin and increased leptin resistance. This finding suggests a possible role of cilia in the leptin pathway, as BBS proteins are required for leptin receptor signaling in mouse BBS models (Rahmouni et al, 2008; Guo and Rahmouni, 2011; Berbari et al, 2013).
McKusick Kaufman Syndrome
McKusick Kaufman syndrome (MKKS; MIM ID#236700) is a rare autosomal recessive disorder characterized by the triad postaxial polydactyly, hydrometrocolpos with vaginal atresia or imperforate hymen and congenital heart disease. In males affected, MKKS syndrome can manifest with hypospadias or cryptorchidism. MKKS is the only gene identified. It is important to discriminate with Bardet-Biedl syndrome which can present also postaxial polydactyly and hidrometrocolps. The main features of BBS patients (obesity, retinal dystrophy and learn disabilities) are age-dependent, so in most cases we cannot certainly diagnose MKS syndrome until childhood (Yewalkar et al, 2013; Son et al 2011;Slavotinek et al, 2000).
Alström Syndrome
Alström syndrome (ALS; MIM ID#203800) is an autosomal recessive disorder characterized by rod-cone dystrophy, obesity, progressive sensorineural hearing impairment, insulin resistance with hyperinsulinemia and dilated cardiomyopathy. In contrast to BBS, ALS syndrome is characterized by relative preservation of cognitive function and the absence of polydactyly. ALS is caused by mutations in ALMS1, which is localized in centrosome and basal body of primary cilia. The location of the proteins encoded by genes NPHP, BBS and ALMS1 highlights the involvement of centrosome in the development of renal cysts, diabetes, obesity, and retinitis pigmentosa.
In both BBS and ALS, truncal obesity develops in childhood and is thought to be linked to hyperphagia (Guo et al, 2011; Minton et al, 2006; Gupta et al, 2009). In ALS, an endocrine phenotype of hyperinsulinemia, insulin resistance and type 2 diabetes are common, while diabetes is also a secondary feature of BBS suggesting that ALMS1 might have a role in β-cell function or peripheral insulin signaling pathways. In Australian studies, obese mice have a spontaneous mutation of the gene responsible for ALS in humans. Although the mice are born at a normal weight, they later exhibit hyperphagia and become obese developing insulin resistance, diabetes and features of metabolic syndrome (Girard et al, 2011; Hearn et al, 2005; Arsov et al, 2006; Romano et al, 2008).
The phenotype of ALS frequently overlaps with BBS, which is thought to be caused by centrosome and/or basal body dysfunction. This dysfunction is a key mechanism in the pathogenesis of obesity, insulin resistance and type 2 diabetes (Hearn et al, 2005).
Orofaciodigital Syndrome Type 1
Orofaciodigital syndrome type 1 (OFDI, MIM ID#311200) is a rare X- linked dominant disorder. Affected males die in utero. The causative gene Ofd1 encodes a protein which is essential in the regulation of cilia and organization of centrosomes. This syndrome is characterized by malformations of the face (frontal bossing, facial asymmetry, hypertelorism and broad nasal bridge), oral cavity (cleft palate and tongue, abnormal dentition) and digital anomalies in girls. OFD1 patients can also have renal cysts and central nervous system abnormalities. (Naiboglu et al, 2012; Coene et al, 2009).
Ciliopathies and Skeletal Defects
The primary cilium plays an essential role in the development of cartilage and bone growth and regulates “hedgehog signaling”. The hedgehog proteins pathway is significantly involved in the differentiation of chondrocytes and osteoblasts in limbs and skeletal axial formation. Recently, it has been shown the role of the primary cilium with mechanotransduction properties in cartilage and bone responding to dynamic fluid flow in cultures being responsible for osteogenesis and bone resorption (Nguyen and Jacobs, 2013; Serra R, 2008).
Most genes involved in skeletal ciliopathies encode proteins responsible for intraflagellar transport and dynein motor, or encode proteins of the basal body. There is a clinical overlap between skeletal ciliopathies and other osteochondrodysplasias which highlights the role of the primary cilium in a broad spectrum of skeletal diseases.
Short-rib polydactyly syndromes (SRPs) are a heterogenous group of recessive disorders characterized by short ribs and limbs, skeletal defects complicated by thoracic hypoplasia, polydactyly and congenital heart defects or renal dysplasia. They are the most severe skeletal ciliopathies with lethality in postnatal life. SRPs have been classified into five subtypes: SRP type I (Saldino-Noonan syndrome, MIM ID#263530), type II (Majewski syndrome, MIM ID#263520), type III (Verma-Naumoff syndrome, MIM ID#263510), type IV (Beemer-Langer syndrome, MIM ID# 269860), and the recently type V (MIM ID#614091). There is a clinical and genetic overlap between five types of SRP and other non-lethal skeletal ciliopathies (Aideen et al, 2013; Huber et al, 2013; Mill et al, 2011). Recently, it has been reported mutation in WD repeat containing protein 34 (WDR34) as causative of SRP type III and severe asphyxiating thoracic dysplasia. WDR 34 has been implicated in the immune response throghough the inhibition of NF-kappa B activation pathway linking immunological processes to skeletal ciliopathies (Huber et al, 2013).
Jeune asphyxiating thoracic dystrophy (JATD, MIM ID#208500) is a rare autosomal recessive chondrodysplasia with skeletal anomalies, primarily shortened ribs and typical pelvis configuration. Abnormalities in ribs lead to a severely constricted thoracic cage and death by respiratory insufficiency. Skeletal abnormalities include a narrow thorax with short ribs, trident acetabular roofs and irregular metaphyseal ends. About 30% of patients also develop renal failure with hepatic fibrosis and retinal involvement less frequently. Mutations have been described in genes IFT80, IFT139/TTC21B, DYNC2H1, IFT 140, WDR19/IFT144, WDR60 and WDR34, genes that encode proteins that are involved in cilia intraflagellar transport (Huber et al, 2013; Aideen et al, 2013; Schmidts et al, 2013; Tüysüz et al, 2009).
Ellis-van Creveld syndrome (EVC, MIM ID#225500) is a recessive chondral and ectodermal dysplasia. EVC is characterized by short ribs and limbs, polydactyly, dysplastic nails and teeth, orofacial abnormalities and cardiovascular malformations. About 60% of EVC patients have heart defects, especially abnormalities of atrial sept. The presence of orofacial malformations such as missing teeth, labio-gingival adherences or maxilar and mandibular anatomic anomalies can help to the diagnosis of this syndrome. Mutations in EVC and EVC2 gene have been identified as causative. Weyers acrofacial dysostosis (EVS, MIM ID# 193530) is an autosomal-dominant syndrome characterized by postaxial polydactyly in addition to anomalies of the lower jaw, dentition and oral vestibule. These syndromes are caused by mutations in the gene encoding EVC proteins, which are located on the basal body of primary cilium and are involved in the hedgehog signaling pathway (Baujat and Le Merrer, 2007).
Sensenbrenner syndrome or cranioectodermal dysplasia (CED, MIM ID#218330) is a rare recessive disorder characterized by dolichocephaly, sagittal craniosynostosis and narrow chest. CED patients can present ectodermal dysplasia in the form of sparse hair and microdontia. Other secondary features are ophthalmologic problems, nephronopthisis and cystic liver disease. Mutations have been reported in IFT122, WDR35, C14orf179 and WDR19; all these genes encode components of intraflagellar transport complex- A (Alazami et al, 2014; Hoffer et al, 2013; Ruiz-Perez and Goodship, 2009).
Ciliopathies and Sense Organs
The primary cilia play a crucial role in vision, olfaction and hearing. Cilia act as photoreceptors in the retina and as mechanoreceptors in the olfactory epithelium and inner ear. The primary cilium dysfunction may lead to alteration of a sense organ in isolation or be involved in the pathogenesis of a complex clinical syndrome.
Retinal Ciliopathies
Photoreceptors in the retina have an inner and outer segment linked by a connecting primary cilium. Photoreceptor disc visual pigments are synthesized exclusively in the inner segment and transported by intraflagellar transport (IFT) along the connecting cilium to the outer segment. Dysfunctional IFT causes perturbation of rhodopsin transport and collapse outer segment leading in blindness (Waters and Beales, 2011).
Several proteins involved in ciliopathic syndromes have been localized in photoreceptor cilium. Retinal degeneration due to ciliary dysfunction is present as part of a wide spectrum of diseases including retinitis pigmentosa, macular degeneration, Leber congenital amaurosis, Senior Loken syndrome and retinal degeneration present in Joubert syndrome, Bardet-Biedl syndrome, Meckel-Gruber syndrome, McKusick-Kaufman syndrome, Alstrom syndrome, primary ciliary dyskinesia, some skeletal ciliopathies and Usher syndrome (Adams et al, 2007).
Mutations in RPGR (retinitis pigmentosa GTPase regulator) are associated with major causes of retinitis pigmentosa including X-linked inheritance and simple forms of RP. RPGR is also involved in primary ciliary dyskinesia and hearing loss. It is localized at the primary cilium of photoreceptors and can interact with several ciliopathic proteins including RPGR-interacting protein I (RPGRIP1), RPGR interacting protein 1-like (RPGRIP1L/NPHP8), NPHP5 and CEP290/NPHP6. RPGR interacting protein 1-like (RPGRIP1L/NPHP8) is also involved in sonic hedgehog signaling and left-right asymetry. Mutations in RPGRIP1L are associated with Joubert syndrome, Meckel-Gruber syndrome and Bardet-Biedl syndrome (Ghosh et al, 2010; Murga-Zamalloa et al, 2009).
Ciliopathies and Inner Ear
Hair cells in the inner ear have mechanosensory receptors that convert vibrations into electrical signals by a deflection of bundles leading to despolarization. Each hair bundle is comprised by a single primary cilium known as kinocilium with “9+2” axoneme pattern flanked by multiple interconnected stereocilia with actin projections. The stereocilia are organized in decreasing height with the longest stereocilia next to the kinocilium. The morphology of hair cells is necessary to a correct functional polarity and mechanosensitivity reception in response to bundle deflection (Schwander et al, 2010).
Kinocilium is thought to play an important role in hair bundle polarity. Recent studies demonstrated that mutations in Bbs-mice and perturbation in intraciliary transport protein Ift88 leads to misoriented hair bundles (Ross et al, 2005; Jones et al, 2008).
Ciliopathies and Olfaction
The olfactory receptor cells are bipolar neurons with a “9+2” pattern. These olfactory cilia lack dynein arms and are therefore immotile. Olfactory receptors, found in the apical region of the olfactory sensory neurons, transduce odor stimuli into changes in neuronal membrane potential by a G-protein mechanism to activate adenyl cyclase type III (Jenkins et al, 2009). The depolarization initiates in response to cAMP binding and is caused by the sequence of 2 currents: an influx of cations (mainly Ca2+) through cyclic nucleotide-gated channel activation and a secondary efflux of Cl− through Ca2+-gated Cl− channel. In this way, olfactory cilia respond to mechanical stimulus that is transduced to an electrical signal.
Reduced olfactory function is present in almost 50% of Bardet-Biedlpatients. The loss of function of BBS proteins causes defects in olfactory cilia structures and leads to olfactory impairment. Mutations in BBS1 ad BBS4 gene and also gene deletion in BBS1, BBS2 and BBS4 in mice lead to impaired olfactory function (Kulaga et al. 2004; Jenkins et al, 2009). Recently, it has been demonstrated that Bbs8 -null mice exhibit olfactory dysfunction as a defect in axon targeting and signaling pathway (Abigail et al, 2011).
Hypomorphic mutations in CEP290/NPHP6 reveal an anosmic dysfunction and retinal degeneration in mouse model. Olfactory dysfunction is due to a defect in localization of olfactory G proteins in primary cilia leading a nonfunctional signaling pathway, despite cilia structure in sensory neurons remains intact (Dyke et al, 2007).
Conclusions
The ciliopathies are a genetically heterogeneous group of clinical entities resulting from dysfunction of motile or sensory cilium. The phenotypic diversity associated with ciliary dysfunction reflects the variability of ciliary protein expression in different cells and tissues. The situation of the cilia in the cell surface facilitates its function as a sensor and transmitter of information between the cell and the extracellular space. Thus, cilia can take on different roles; for example, modulating the flow direction in the extracellular embryonic node or mediating intracellular calcium signaling in the presence of renal flow. Cilia dysfunction encompasses a large group of disorders ranging from primary ciliary dyskinesia changes in left-right laterality, retinitis pigmentosa, sensorineural deafness and oncogenesis. As a result, ciliopathies can present with different phenotypes, although they often share renal, bone, ocular and central nervous system defects. These groups of disorders are also genetically heterogeneous and it remains possible that mutation of a single gene results in different phenotype according to the time for action in the embryo. Ciliopathies types based on the participation of the motile or primary cilium not support a clear differentiation between these disorders, therefore it could be more practical to know the age of onset of symptoms, their lethality and the possibility of renal or bone involvement. Recently, cilia dysfunction has been implicated in the pathogenesis of other conditions apparently unrelated to the recognized ciliopathies including obesity, hypertension and diabetes. These findings may offer new targets for therapeutic intervention.
Acknowledgments
ISCIII CIBERER is an initiative of ISCIII Ministry of Economy and Competitiveness of Spain.
Reference
1. Adams, N.A., Awaidein, A. and Tomas, H.S. (2007) “The retinal ciliopathies,” Ophtalmic Genetics, 28 (3) 113-125.
2. Afzelius, B.A. (1995) “Situs inversus and ciliary abnormalities. What is the connection?,” International Journal of Developmental Biology, 39 (5) 839-844.
3. Aideen, M., Mc Inerney-Leo, Schmidts, M., Wicking, C. (2013)”Short-Rib Polydactyly and Jeune Syndromes Are Caused by Mutations in WDR60,” American Journal of Human Genetics, 93 (3) 515-523.
4. Alazami, A.M., Seidahmed, M.Z., Alzahrani, F., Mohammed, A.O. and Alkuraya, F.S. (2014) “Novel IFT122 mutation associated with impaired ciliogenesis and cranioectodermal dysplasia,” Molecular Genetics and Genomic Medicine, 2 (2) 103-106.
5. Ansley, S.J., Badano, J.L., Blacque, O.E., Hill, J., Hoskins, B.E., Leitch ,C.C. et al. (2003) “Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome,” Nature, 425 (6958) 628-633.
6. Arsov, T., Silva, D.G., O´Bryan, M.K., Sainsbury, A., Lee, N.J., Kennedy, C. et al. (2006) “Fat aussie–a new Alström syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis,” Molecular Endocrinology, 2006; 20 (7) 1610-1622.
7. Arts, H.H. and Knoers, N.V. (2013) “Current insights into renal ciliopathies: what can genetics teach us?,” Pediatric Nephrology, 28 (6) 863-874.
8. Ashe A., Butterfield N.C., Town L., Courtney A.D., Cooper A.N., Ferguson C. et al (2012) “Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies,” Human Molecular Genetics, 21 (8) 1808-1823.
9. Bandano, J.L., Mitsuma, N., Beales, P.L. and Katsanis N. (2006) “The ciliopathies: and emerging class of human genetic disorders,” Annual Review of Genomics and Human Genetics, 22 125-148.
10. Baker, A.R., Thomas, R. and Dawe, H.R. (2014) “Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development,” Organogenesis, 10 (1) 96-107.
11. Baker, K. and Beales, P.L. (2009). “Making sense of cilia in disease: the human ciliopathies,” American Journal of Medical Genetics Part C Seminars in Medical Genetics, 151C (4) 281-295.
12. Basten, S.G. and Giles, R.H. (2013) “Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis,” Cilia, 2 (1) 6.
13. Baujat, G. and Le Merrer, M. (2007) “Ellis-Van Creveld syndrome,” Orphanet Journal of Rare disease ,2:27.
14. Beales, P.L., Eleioglu, N., Woolf, A.S., Parker, D. and Flinter, F.A. (1999) “New criteria for improved diagnosis of Bardet-Biedl syndrome: results of apopulation survey,” Journal of Medical Genetics, 36 (6) 437-446.
15. Berbari, N.F., O’Connor A.K., Haycraft, C.J. and Yoder, B.K. (2009) “The primary cilium as a complex signaling center,” Current Biology, 19 (13) R526-R535.
16. Berbari, N.F., Pasek, R.C., Malarkey, E.B., Yazdi, S.M., McNair, A.D., Lewis, W.R. et al. (2013) “Leptin resistance is a secondary consequence of the obesity in ciliopathy mutant mice,” Proceedings of the National Academy of Sciences of the United States of America, 110 (19) 7796-7801.
17. Bisgrove, B.W. and Yost, H.J. (2006) “The roles of cilia in developmental disorders and disease,” Development, 133 (21) 4131-4143.
18. Cardenas-Rodriguez, M. and Badano, J.L. (2009) “Ciliary biology: understanding the cellular and genetic basis of human ciliopathies,” American Journal of Medical Genetics Part C Seminars in Medical Genetics, 151C (4) 263-280.
19. Coene, K.L., Roepman, R., Doherty, D., Afroze, B., Kroes, H.Y., Letteboer, S.J. et al. (2009) “OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-Encoded Lebercilin,” American Journal of Human Genetics, 85 (4) 465-481.
20. Davenport, J.R. and Yoder, B.K. (2008) “An incredible decade for the primary cilium: a look at a once-forgotten organelle,” American Journal of Physiology – Renal Physiology, 289 (6) F1159-F1169.
21. Davis, E.E. and Katsanis, N. (2012) “The ciliopathies: a transitional model into systems biology of human genetic disease,” Current Opinion in Genetics & Development, 22(3) 290-303.
22. Dawe, H.R., Smith, U.M., Cullinane, A.R., Gerrelli, D., Cox, P., Badano, J.L. et al. (2007) “The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation,” Human Molecular Genetics, 16 (2) 173-186.
23. den Hollander, A.I., Roepman, R., Koenekoop, R.K. and Cremers, F.P. (2008) “Leber congenital amaurosis: genes, proteins and disease mechanisms,” Progress in Retinal Eye Research, 27 (4) 391-419.
24. Dowdle, W.E., Robinson, J.F., Kneist, A., Sirerol-Piquer, M.S., Frints, S.G, Corvit, K.C. et al. (2011) “Disruption of a Ciliary B9 protein Complex causes Meckel Syndrome,” American Journal of Human Genetics, 89 (1) 94-110.
25. Eccles, M.R. and Stayner, C.A. (2014) “Polycystic kidney disease – where gene dosage counts,” Biomedical Review Journals-F1000 Prime Reports, 6:24.
26. Egeberg, D.L., Lethan, M., Manguso, R., Schneider, L., Awan, A., Jorgensen, T. et al. (2012) “Primary cilia and aberrant cell signaling in epithelial ovarian cancer,” Cilia, 1 (1) 15.
27. Eggenschwiler, J.T. and Anderson, K.V. (2007) “Cilia and developmental signaling,” Annual Review of Cell and Developmental Biology, 23: 345-373.
28. Esteban, M.A., Harten, S.K., Tran, M.G. and Maxwell, P.H. (2006) “Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein,” Journal of the American Society of Nephrology, 17 (7) 1801-1806.
29. Estrada-Cuzcano, A., Roepman, R., Cremers, F.P., Den Hollander, A.I. and Mans, D.A. (2012) “Non-syndromic retinal ciliopathies: translating gene discovery into therapy,” Human Molecular Genetics, 21 (R1) R111-R124.
30. Feistel, K. and Blum, M. (2006) “Three types of cilia including a Novel 9+4 axoneme on the Notochordal Plate of Rabbit Embryo,” Developmental Dynamics, 235 (12) 3348-3358.
31. Ferkol, T.W. and Leigh, M.W. (2012) “Ciliopathies: the central role of cilia in a spectrum of pediatric disorders,” Journal of Pediatrics, 160 (3) 366-371.
32. Fliegauf, M., Benzing, T. and Omran, H. (2007) “When cilia go bad: cilia defects and ciliopathies,” Nature Reviews Molecular Cell Biology, 8 (11) 880-893.
33. Furuya, M. and Nakatani, Y. (2013) “Birt-Hogg-Dubé syndrome: clinicopathological features of the lung,” Journal of Clinical Pathology, 66 (3) 178-186.
34. Garcia-Gonzalo, F.R. and Reiter, J.F. (2012) “Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access,” Journal of Cell Biology, 197 (6) 697-709.
35. Gerdes, J.M., Davis, E.E. and Katsanis, N. (2009) “The vertebrate primary cilium in Gudevelopment, homeostasis, and disease,” Cell, 137 (1) 132-145.
36. Girard, D. and Petrovsky, N. (2011) “Alström syndrome: insights into the pathogenesis of metabolic disorders,” Nature Reviews Endocrinology, 7(2):77-88.
37. Gosh, A.K., Murga-Zamalloa, C.A., Chan, L., Hitchcock, P.F., Swaroop, A. and Khanna, H. (2010) “Human retinopathy-associated ciliary protein retinitis pigmetosa GTPase regulator mediates cilia-dependent vertebrate development,” Human Molecular Genetics, 19 (1): 90-98.
38. Gradilona, S.A., Masyuk, A.I., Splinter, P.L., Banales, J.M., Huang, B.P., Tietz, PS et al. (2007) “Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion,” Proceedings of the National Academy of Sciences of the United States of America, 104 (48) 19138-19143.
39. Gunay-Aygun, M. (2009) “Liver and kidney disease in ciliopathies,” American Journal of Medical Genetics Part C Seminars in Medical Genetics, 151C (4) 296-306.
40. Guo, D.F. and Rahmouni K. (2011) “Molecular basis of the obesity associated with Bardet-Biedl syndrome,” Trends in Endocrinology and Metabolism, 22 (7) 286-293.
41. Gupta, P.S., Prodromou, N.V. and Chapple, J.P. (2009) “Can faulty antennae increase adiposity? The link between cilia proteins and obesity,” Journal Endocrinology, 203 (3) 327-336.
42. Hearn, T., Spalluto, C., Phillips, V.J., Renforth, G.L., Copin, N., Hanley, N.A.et al. (2005) “Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes,” Diabetes 54 (5) 1581-1587.
43. Hildebrandt, F., Attanasio, M. and Otto, E. (2009) “Nephronophthisis: disease mechanisms of a ciliopathy,” Journal of the American Society of Nephrology, 20 (1) 23-35.
44. Hildebrandt, F., Benzing, T. and Katsanis, N. (2011) “Ciliopathies,” New England Journal of Medicine, 364 (16):1533-1543.
45. Hoffer, J.L., Fryssira, H., Konstantinidou, A.E., Ropers, H.H. and Tzschach, A. (2013) “Novel WDR35 mutations in patients with cranioectodermal dysplasia (Sensenbrenner syndrome),” Clinical Genetics, 83 (1) 92-95.
46. Huber, C., Wu, S., Kim, A.S., Sigaudy, S., Sarukhanov, A., Serre, V. et al. (2013) “WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-κB pathway in cilia,” American Journal of Human Genetic, 93 (5) 926-931.
47. Hunkapiller, J., Singla, V., Seol, A. and Reiter, J.F. (2011) “The ciliogenic protein Oral-Facio-Digital 1 regulates the neuronal differentiation of embryonic stem cells,” Stem Cells Development, 20 (5) 831-841
48. Hurd, T.W. and Hildebrant, F. (2011) “Mechanisms of Nephronophthisis and related ciliopathies,” Nephron Experimental Nephrology, 118 (1) e9-e14
49. Icardo, J.M., García, J.M. and Ros, M.A. (2002) “Congenital heart disease, heterotaxia and laterality,” Revista Española de Cardiología, 55 (9) 962-974.
50. Jan, A. (2013) “Mutations in CIB2 calcium and integrin-binding protein disrupt auditory hair cell calcium homeostasis in Usher syndrome type 1J and non-syndromic deafness DFNB48,” Clinical Genetics, 83 (4) 317-318.
51. Jenikins, P.M., McEwen, D.P. and Martens, J.R. (2009) “Olfactory cilia: linking sensory cilia function and human disease,” Chemical Senses, 34 (5):451-464.
52. Jones, C., Roper, V.C., Foucher, I., Qian, D., Banizs, B., Petit, C. et al. (2008) “Ciliary proteins link basal body polarization to planar cell polarity regulation,” Nature Genetics, 40 (1) 69-77.
53. Keats, B.J. and Corey, D.P. (1999) “The usher syndromes,” American Journal of Medical Genetics, 89 (3) 158-166.
54. Kennedy, M.P., Omran, H., Leigh, M.W., Dell, S., Morgan, L., Molina, P.L. et al. (2007) “Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia,” Circulation, 115 (22) 2814-2821.
55. Kim, S. and Dynlacht, B.D. (2013) “Assembling a primary cilium,” Current Opinion in Cell Biology, 25 (4) 506-511.
56. Koenekoop, R.K. (2004) “An overview of Leber congenital amaurosis: a model to understand human retinal development,” Survey Ophthalmology 49 (4) 379-398.
57. Kulaga, H.M., Leitch, C.C. Eichers, E.R., Badano, J.L., Lesemann, A., Hoskins, B.E. et al. (2004) “Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse,” Nature Genetics, 36 (9) 994-998.
58. Leigh, M.W., Pittman, J.E., Carson, J.L., Ferkol, T.W., Dell, S.D., Davis, S.D. et al (2009) “Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome,” Genetics in Medicine, 11 (7) 473-487.
59. Liu Y.P., Tsai I.C., Morleo M., Oh E.C., Leitch C.C., Massa F, et al. (2014) “Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators,” Journal of Clinical Investigation, 124 (5) 2059-2070.
60. Malicki, J. (2012) “Who drives the ciliary highway?,” Bioarchitecture, 2 (4) 111-117.
61. Marshall, W.F. and Nonaka, S. (2006) “Cilia: tuning in to the cell’s antenna,” Current Biology, 16 (15) R604-R614.
62. Masyuk, A.I., Gradilone, S.A., Banales, J.M., Huang, B.Q., Masyuk, T.V., Lee, S.O. et al. (2008) “Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors,” American Journal of Physiology – Gastrointestinal and Liver Physiology, 295 (4) G725-GT34.
63. Martinez-Frías, M.L., Cuevas, L. and Bermejo-Sanchez, E. (2012) “Análisis clínico y epidemiológico de los recién nacidos con defectos congénitos registrados en el ECEMC: Distribución por etiología y por grupos étnicos,” Boletin ECEMC: Revista de dismorfología y epidemiología, VI (2) 18-55.
64. McEwen ,D.P., Koenekoop, R.K., Khanna, H., Jenkins, P.M., Lopez, I., Swaroop, A. and Martens, J.R. (2007)” Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons,” Proceedings of the Natural Academy of Sciences of the United States of America,104 (40) 15917-15922.
65. M´hamdi, O., Ouertani, I. and Chaabouni-Bouhammed, H. (2014) “Update on the genetics of Bardet-Biedl syndrome,” Molecular Syndromology 5 (2) 51-56.
66. Mill, P., Lockhart, P.J., Fitzpatrick, E., Mountford, H.S., Hall, E.A., Rejins, E.A. et al. (2011) “Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis,” American Journal of Human Genetic, 88 (4) 508-515.
67. Minton, J.A., Owen, K.R., Ricketts, C.J., Crabtree, N., Shaikh, G., Ehtisham, S. et al. (2006) “Syndromic obesity and diabetes: changes in body composition with age and mutation analysis of ALMS 1 in 12 United Kingdom kindreds with Alström syndrome,” Journal of Clinical Endocrinology and Metabolism, 91 (8) 3110-3116.
68. Mockel, A., Perdomo, Y., Stutzmann, F., Letsch, J., Marion, V. and Dollfus H.(2011) “Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies,” Progress in Retinal and Eye Research, 30 (4) 258-274.
69. Murga-Zamalloa, C.A., Swaroop, A. and Khanna, H. (2009) “RPGR-containing protein complexes in syndromic and non-syndromic degeneration due to ciliary dysfunction,” Journal of genetics, 88 (4) 399-407.
70. Naiboglu B., Oysu C. and Gokceer T. (2012) “Orofaciodigital syndrome,” Ear, Nose & Throat Journal, 91 (1) E8-E9.
71. Nguyen, A.M. and Jacobs, C.R. (2013) “Emerging role of primary cilia as mechanosensors in osteocytes,” Bone, 54 (2) 196-204.
72. Pan, J., Wang, Q. and Snell, W.J. (2005) “Cilium-generated signaling and cilia-related disorders,” Laboratory Investigation, 85 (4) 452-463.
73. Parelkar, S.V., Kapadnis, S.P., Sanghvi, B.V., Joshi, P.B., Mundada, D. and Oak, S.N. (2013) “Meckel-Gruber syndrome: A rare and lethal anomaly with review of literature,” Journal of Pediatrics Neurosciences, 8 (2) 154-157.
74. Perrault, I., Delphin, N., Hanein, S, Gerber, S., Dufier, J.L., Rocher, O. et al. (2007) “Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype,” Human Mutation, 28 (4) 416.
75. Popatia, R., Haver, K. and Casey, A. (2014) “Primary Ciliary Dyskinesia: An Update on New Diagnostic Modalities and Review of the Literature,” Pediatric Allergy, Inmunology and Pulmonology Journal, 27 (2) 51-59.
76. Quilan, R.J., Tobin, J.L. and Beales, P.L. (2008) “Modeling ciliopathies: Primary cilia in development and disease,” Current Topics in Developmental Biology, 84 249-310.
77. Rachel, R.A., Li, T. and Swaroop, A. (2012) “Photoreceptor sensory cilia and ciliopathies: focus on CEP290, RPGR and their interacting proteins”, Cilia, 1 (1) 22.
78. Rahmouni ,K., Fath, M.A., Seo, S., Thendens, D.R., Berry, C.J., Weiss R. et al. (2008) “Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome,” Journal of Clinical Investigation, 118 (4) 1458-1467.
79. Roepman, R. and Wolfrum, U. (2007) “Protein networks and complexes in photoreceptor cilia,” Subcellular Biochemistry, 43 209-235.
80. Romani, M., Micalizzi, A., Kraoua, I., Dotti, MT., Cavallin, M., Sztriha, L., et al. (2014) “Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome,” Orphanet Journal of Rare Diseases, 9 72.
81. Romani, M., Micalizzi, A. and Valente E.M. (2013) “Joubert syndrome: congenital cerebellar ataxia with the molar tooth,” Lancet Neurology, 12 (9) 894-905.
82. Romano, S., Milan, G., Veronese, C., Collin, G.B., Marshall, J.D., Centobene, C. et al. (2008) “Regulation of Alström syndrome gene expression during adipogenesis and its relationship with fat cell insulin sensitivity,” International Journal of Molecular Medicine 2008 21 (6) 731-736.
83. Ronquillo, C.C., Bernstein, P.S. and Baehr, W. (2012) “Senior-Løken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis,” Vision Research, 75 88-97.
84. Ross, A.J., May-Simera, H., Eichers, E.R., Kai, M., Hill, J., Jagger D.J., et al (2005) “Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates,” Nature Genetics, 37 (10) 1135-1140.
85. Ruiz-Perez, V.L. and Goodship, J.A. (2009) “Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to hedgehog ligands,” American Journal of Medical Genetics Part C Seminars in Medical Genetics, 151C (4) 341-351.
86. Sattar, S. and Gleeson, J.G. (2011) “The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders,” Developmental Medicine and Child Neurology, 53 (9) 793-798.
87. Schmidts, M., Vodopiutz, J., Chirstou-Savina, S., Cortés, C.R., McInerney-Leo, A.M., Emes, R.D. et al. (2013) “Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy,” American Journal of Human Genetics, 93 (5) 932-944.
88. Schwander, M., Kachar, B. and Müller U. (2010) “The cell biology of hearing,” Journal Cell Biology, 190 (1) 9-20.
89. Serra R. (2008) “Role of Intraflagellar Transport and Primary Cilia in Skeletal Development”, Anatomical Record, 291 (9) 1049-1061.
90. Sharma, N., Berbari, N.F. and Yoder, B.K. (2008) “Ciliary dysfunction in developmental abnormalities and diseases,” Current Topics in Developmental Biology, 85 371-427.
91. Shorbagi, A. and Bayraktar, Y. (2010) “Experience of a single center with congenital hepatic fibrosis: a review of the literature,” World Journal Gastroenterology, 16 (6) 683-690.
92. Singla, V. and Reiter, J.F. (2006) “The primary cilium as the cell’s antenna: signaling at a sensory organelle,” Science, 313 (5787) 629-633.
93. Slavotinek, A.M. and Biesecker, L.G. (2000) “Phenotypic overlap of McKusick-Kaufman syndrome with bardet-biedl syndrome: a literature review,” American Journal of Human Genetics, 95 (3) 208-215
94. Son, S.H., Kim, Y.J., Kim, E.S., Kim, E.K., Kim, H.S. and Choi, J.H. (2011) “A case of McKusick-Kaufman syndrome,” Korean Journal of Pediatrics, 54 (5) 219-223.
95. Tadenev, A.L., Kulaga, H.M., May-Simera, H.L., Kelley, M.W., Katsanis, N. and Reed, R.R. (2011) “Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting,” Proceedings of the Natural Academy of Sciences of the United States of America, 108 (25) 10320-10325.
96. Tammachote, R., Hommerding, C.J., Sinders, R.M., Miller, C.A., Czarnecki, P.G., Leightner, A.C. et al. (2009) “Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3,” Human Molecular Genetics, 18 (17) 3311-3323.
97. Toftgård, R. (2009). “Two sides to cilia in cancer,” Nature Medicine, 15 (9) 994-996.
98. Torres, V.E. and Harris, P.C. (2009) “Autosomal dominant polycystic Kidney disease: the last 3 years,” Kidney International, 76 (2) 149-168.
99. Tüysüz, B., Bariş, S., Aksoy, F., Madazli, R., Ungür, S. and Sever, L. (2009) “Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: Evaluation and classification of 13 patients,” American Journal of Medical Genetics Part A, 149A (8) 1727-1733.
100. Valente, E.M., Dallapiccola, B. and Bertini, E. (2013) “Joubert syndrome and related disorders,” Handbook of Clinical Neurology, 113: 1879-1888.
101. Wang, H., den Hollander, A.I., Moayedi, Y., Abulimiti, A., Li, Y., Collin, R.W. et al. (2009) “Mutations in SPATA 7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa,” American Journal of Human Genetics, 84 (3) 380-387.
102. Ware, S.M., Aygun, M.G. and Hildebrant, F. (2011) “Spectrum of clinical diseases caused by disorders of primary cilia,” Proceedings of the American Thoracic Society, 8 (5) 444-450.
103. Waters, A.M. and Beales, P.L. (2010) “Ciliopathies: and expanding disease spectrum,” Pediatric Nephrology, 26 (7) 1039-1056.
104. Wolf, M.T. and Hildebrandt, F. (2011) “Nephronophthisis,” Pediatric Nephrology, 26 (2) 181-194.
105. Wong S.Y., Seol A.D., So P.L., Ermilov A.N., Bichakjian C.K., Epstein E.H., Duglosz A.A. and Reiter J.F. (2009) “Primary cilia can both mediate and suppress hedgehog pathway dependent tumorigenesis,” Nature Medicine, 15 (9) 1055-1061.
106. Yewalkar, S.P., Yadav, V.K. and Khadse, G.J. (2013) “The McKusick-Kaufman hydrometrocolpos-polydactyly syndrome: A rare case report,” Indian Journal of Radiology and Imaging, 23 (2) 183-185.
107. Yuan, S. and Sun, Z. (2013) “Expanding horizons: ciliary proteins reach beyond cilia,” Annual Review of Genetics, 47 353-76.
108. Zariwala, M.A., Knowles, M.R. and Leigh M.W. (2007) “Primary Ciliary Dyskinesia,” In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2013. Available from http://www.ncbi.nlm.nih.gov/books/NBK1122/