Euphrosyni Koutsouraki, Dimitrios Michmizos, Thalia Kalatha and Stavros Baloyannis
1st Department of Neurology, Aristotle University, AHEPA Hospital, Thessaloniki, Greece
Volume 2013 (2013),
Article ID 477527,
International Journal of Case Reports in Medicine,
13 pages,
DOI: 10.5171/2013.477527
Received date: 10 February 2013; Accepted date: 13 March 2013; Published date: 24 April 2013
Academic Editor: Liang Fu Zhou
Cite this Article as:
Euphrosyni Koutsouraki, Dimitrios Michmizos, Thalia Kalatha and Stavros Baloyannis (2013), "A Rare Case of Extensive Cavernous Hemangioma of the Brainstem Combined with Multiple Pathological and Anatomical Abnormalities," International Journal of Case Reports in Medicine, Vol. 2013 (2013), Article ID 477527, DOI: 10.5171/2013.477527
We present the case of a 14-year-old Caucasian female, with intracranial hemorrhage due to an extensive brainstem cavernous hemangioma, asymptomatic until the date of her admission. Further investigation revealed multiple brain cavernous hemangiomas with two supratentorial hemangiomas demonstrating signs of past bleeding, a cervical vertebral dysplasia (Klippel-Feil type II) and nephrolithiasis, in addition to her history of polycystic ovaries and impaired glucose tolerance, all together presenting a novel clinical combination. We are describing the management of such a patient, according to our current knowledge, inquiring into the possible connections between the conditions detected, as well as, discussing the questions that arise from this rare case.
Cerebral cavernous hemangiomas (CCHs) represent the most common central nervous system vascular lesions in children, even if their incidence is 4 times lower than that of the adult population. The natural history of pediatric CCHs seems to be more aggressive than in adult patients; these lesions have higher rates of growth and hemorrhage, larger dimensions and often atypical radiological pictures at diagnosis. (Amirjamshidi and Abbassioun, 2000; Nieto et al, 2003).
Cerebral cavernous malformations (CCMs) sometimes called cavernous angiomas or cavernomas are well-circumscribed, lobulated, raspberry-like lesions. CCMs consist of a compact mass of thin-walled, dilated vessels lined by a single layer of endothelium. The vessels can be adjacent to each other or separated by fibrous connective tissue and may exhibit variable hyalinization. The vascular walls lack an organized elastic lamina and tend to be devoid of smooth muscle cells as well. There is usually evidence of a previous hemorrhage, thrombosis from stagnant blood flow, and the lesions may be calcified. In contrast to arteriovenous malformations, intervening parenchymal tissue is scarce or absent and their blood supply is obtained from very small low-flow blood vessels in the brain. Cavernous malformations can occur anywhere in the body, but usually produce serious signs and symptoms only when they occur in the central nervous system (brain and spinal cord). (Del Curling et al, 1991)
Both sporadic and familial forms have been identified. The pattern of inheritance of the familial form is autosomal dominant, affecting mainly patients of Hispanic-American origin. Familial cases are characterised by the presence of multiple lesions, whereas sporadic cases usually have a single lesion (Rigamonti et al., 1988). In familial CCMs, extraneurological localizations are detected in a small proportion and they include retinal and cutaneous cavernomas. Retinal cavernomas occur in 5% of patients with familial type CCMs which are generally stable and uneventful (Moffat et al., 1988) Also, another distinct ocular lesion, choroidal haemangioma, has been described (Sarraf et al., 2000). The cutaneous lesions involved in familial CCM include bluish nodules, cherry angiomas, capillary vascular anomalies, and eruptive multiple angiokeratomas, while hyperkeratotic cutaneous capillary venous malformations are the most characteristic type of lesion and is never present in the general population or in sporadic CCM (Labauge et al., 1999). In addition to these localizations, very occasionally other organs may be affected, as cited by Wood (1957) in a case with multiple cerebral cavernomas and cardiac, renal and cutaneous localizations that were anatomorphopathologically proven to be capillary malformations similar to the cerebral ones.
Klippel-Feil is a syndrome described in 1912 and characterized by short neck, low hairline and limited cervical motion (Klippel M, Feil A, 1912); however, fewer than 50% of the patients with cervical spine congenital defects demonstrate all three signs. There is a constellation of associated conditions related to Klippel- Feil syndrome. These include congenital scoliosis, Sprengel’s deformity, renal disorders (more than one third), synkinesis (mirror motions), congenital heart disorders, brain stem abnormalities, occipital cephalocele, Chiari malformation I, syndactyly and hypoplastic thumb, hearing loss, juvenile rheumatoid arthritis and rheumatoid spondylitis (Tracy et al., 2004; Hensinger et al., 1974, Karaman, A and Kahveci, H, 2011). The actual incidence of Klippel-Feil syndrome (KFS) is unknown due to the fact that there never was any large scale study done to determine this, but it is estimated to be 1:42.000 newborns worldwide. In addition, females seem to be affected slightly more often than males (Gunderson et al., 1967). Even though, a great number of associated anomalies have been described with KFS, the coexistence of multiple cavernomatosis has never been noted in the literature. Only cases of anomalous venous connections, usually pulmonary (Miyamoto et al., 1971) have been mentioned.
Case Report
A 14-year-old Caucasian female was admitted to our clinic suffering from sudden occipital headache. Nausea, vomiting, dysarthria, diplopia, drowsiness and weakness of the right upper and lower extremity was added an hour after admission. Neurological examination demonstrated paresis of the right III, V and XII cranial nerves, paresis of the left VI, VII and VIII cranial nerves and right pyramidal hemiparesis.
The medical history of the patient revealed a laborious childbirth due to short umbilical cord resulting in acute superficial head hematoma, arthritis at the right knee joint and obstetric paralysis of upper left extremity. The last two years she was under medical supervision for polycystic ovarian syndrome and glucose intolerance.
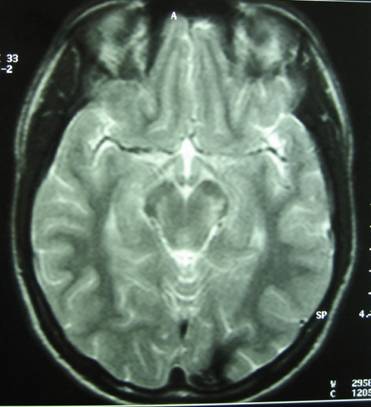
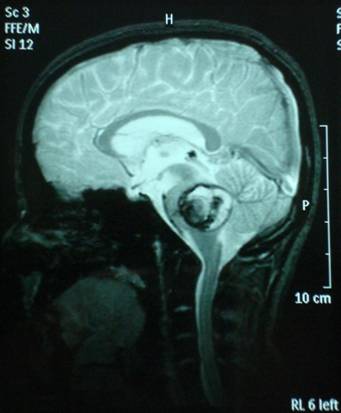
The admission day brain CT revealed the presence of inconsistent hemorrhagic material at the region of the pons with occipital presence of accompanying subarachnoid hemorrhage (Fig.1).
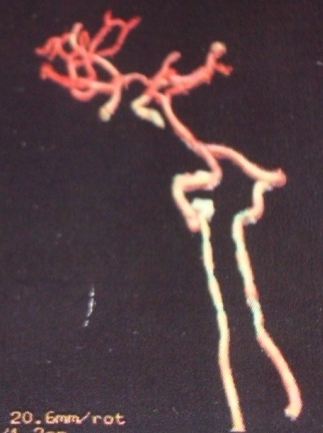
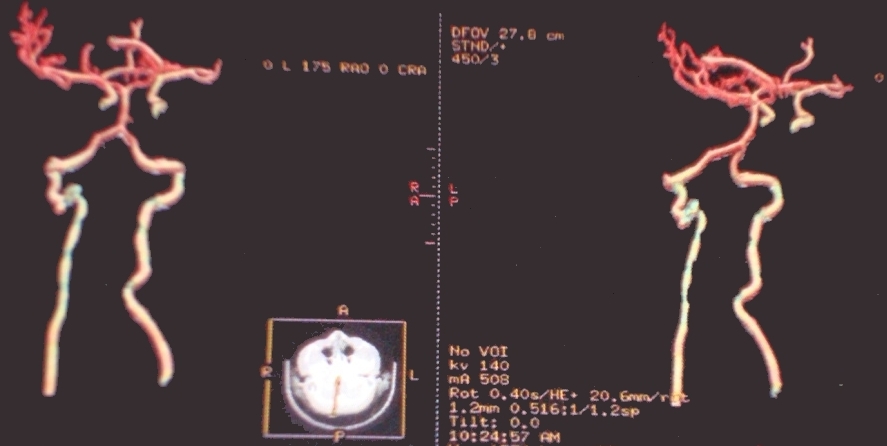
The brain and cervical spinal cord CT angiographies revealed no arteriovenous type damage (Fig 2, 3).
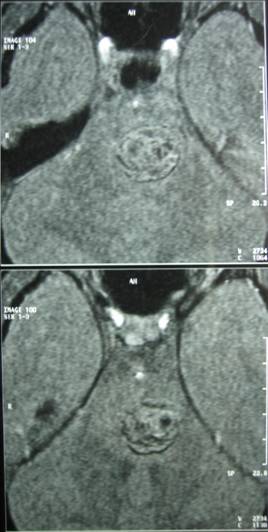
The brain MRI-MRA-MRV showed that the pons had hemorrhaged internally with peripheral deposits of hemoferritin (characteristic of a CCM) (Fig.4).
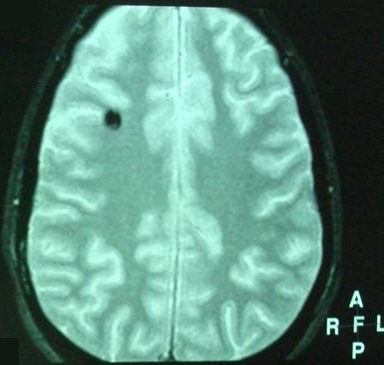
Hemoferritin deposits were also present at two other cavernous angiomas, a) occipitally (L), and b) in the dome of the parietal lobe (R) with characters consistent of previous hemorrhage (Fig 5, 6).
Based on the classification of Zabramski et al. (1994), there is a typical type II cavernoma in the brainstem which is represented by association in T2 of a hyper and hyposignal, surrounded of a crown of hyposignal corresponding to the breakdown products of heamoglobin, known as “popcorn” or “niche of bee” image. Also, two type 3 cavernomas are visible, which corresponds to a visible malformation in the form of a hyposignal in T1 and T2 images.
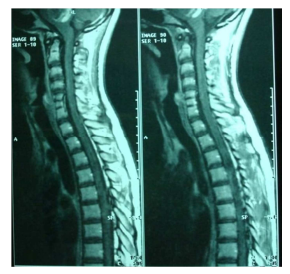
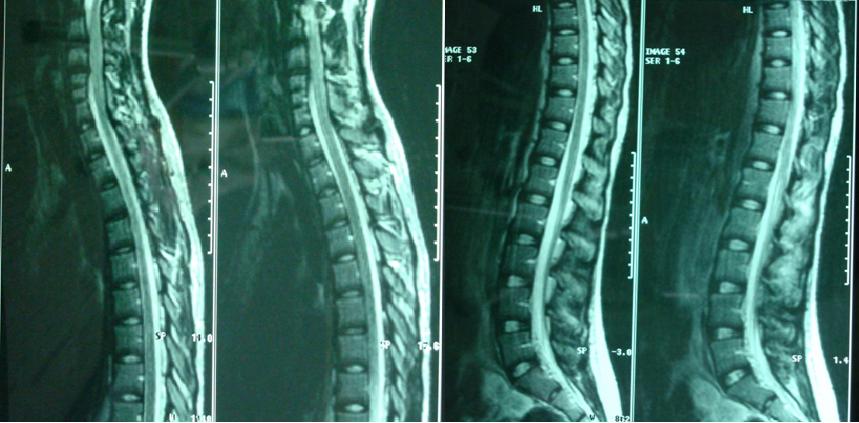
In the spinal cord MRI there was disruption of the normal curvature of the cervical spinal cord region and dysplastic configuration of the C5, C6 and C7 vertebral bodies whose height was diminished (congenital type configuration). The spinal cord at the cervical spine region progressed under narrow tube conditions at the levels of C3-C4 down to C7-T1 (9/10/12/12/10mm vs. 14mm) (Fig. 7, 8).
Thoracic and lumbar spinal cord was normal (Fig. 9). The image of the spine is consistent with Klippel Feil type II, according to Feil’s classification of the malformation based on the site and extend of cervical fusion (MacEwen, 1975). Type I corresponds to patients with extensive cervical and upper thoracic fusion. Type II to patients with one or two cervical interspace fusions, which is the most usual and asymptomatic form of the anomaly, with the fusion between C5-C6 thought to be autosomal recessive (Gunderson et al., 1967). In type III patients have malformations of both cervical and lower thoracic or lumbar spine.
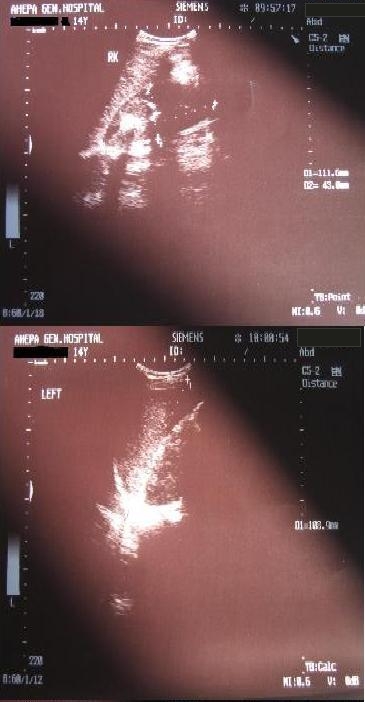
Fundus examination was normal with absence of malformations. Skin examination was also normal, without any evident lesions. The girl did not have a low hairline, neither a short neck. In the examination there was reduced cervical motion (concurrent subarachnoid hemorrhage). In the differential diagnosis some metastases, especially of renal origin that could resemble to the brain MRI image, or syndromes like von Hippel Landau and hemangioblastomas or syndromes with arteriovenous malformations (AVMs), as hereditary hemorrhagic telangiectasia, were thought upon but the MRI image was quite typical for cavernomas and the negative angiographic image excluded developmental venous malformations or AVMs. To exclude other localizations and other pathology ultrasound of the upper and lower abdomen was performed. Liver, bile vessels, gallbladder, pancreas, bladder and uterus were unimpaired. The lower abdomen ultrasound showed bilateral nephrolithiasis and bilateral distention of the pyelocalycal system, especially on the left, as well as of the ipsilateral ureter (Fig. 10).
There was a significant amount of free liquid in the Douglas space. The left ovary was depicted with multiple cystic formations of various sizes. Hormonal examination was normal. Due to the patient’s neonatal history which included arthritis, a full immunological examination was deemed necessary and which yielded no abnormal results, with the exception of a small decrease in the levels of complement component 3.
Due to the hemangioma location (brainstem), size, multiplicity and the absence of seizures or progressive neurological deficits, it was decided for the patient to be treated conservatively.
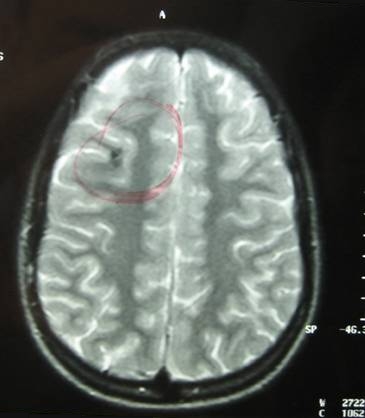
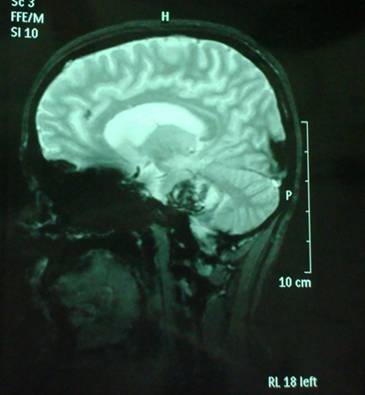
A follow-up brain MRI was performed one month after the onset of the symptoms and it confirmed the presence of an extensive cavernous hemangioma in the brainstem, still not fully absorbed. (Fig. 11, 12, 13).
After two months of conservative treatment the clinical evaluation showed the patient improving. No seizures occurred. Since the patient did not have a positive family history for cavernomatosis or epilepsy, a diagnosis of sporadic cavernomatosis was reached. Sporadic cases usually show a single lesion on both TSE and gradient-echo sequences, have no relative with the same condition and do not carry a CCM gene mutation (Verlaan et al., 2004). However, some cases have multiple MRI lesions although they do not have any known clinically affected relative and present therefore as sporadic cases. In symptomatic sporadic cases with multiple CCM screening for CCM genes may identify a mutation in around 50% of cases. This information will not change the patient’s management but may be useful for genetic counselling. However, the patient should be informed that a negative test does not exclude a genetic cause (Labauge et al., 2007). Genetic screening for KRIT1, KRIT2, KRIT3 was proposed for examination and declined by family.
In our case, the young patient relocated 4 years after admission without leaving a forwarding address, thus a follow-up MRI scan has proven very difficult to manage so far. So, the question remains whether the cavernomas were altered in character or there are new ones. During the last clinical evaluation (2 years after admission) the patient was unimpaired and the girl did not complain of headaches, blurred vision or other neurological symptoms in the above-mentioned period.
Discussion
“Sporadic” Cases with Multiple Lesions
Studies that were performed before the identiï¬cation of CCM genes in a series of 22 consecutive sporadic cases with multiple lesions showed that 75% of them were affected with a hereditary form of the disease with incomplete penetrance because one of their two asymptomatic parents showed lesions on MRI (Labauge et al., 1998). These patients are true familial genetic cases. The remaining 25% of patients whose biological parents had a normal TSE/gradient-echo brain was divided into two groups: in the first group were the ones carrying a de novo mutation in one of the three CCM genes whereas in the second the ones that did not carry any mutation of these genes. To explain the second group of cases researchers suggested a somatic mosaicism of a de novo mutation that occurred during gestation and is not detectable in DNA extracted from peripheral blood cells. Other hypotheses include the possibility of mutations located outside CCM coding exons, large deletions or duplications which would not have been detected by the strategies used so far, and the possibility of an unidentiï¬ed gene (Denier et al., 2004).
Genetics and CCM
As mentioned, CCMs can occur sporadically or as an autosomal dominant condition with variable expression and incomplete penetrance. Familial forms have been linked to three chromosomal loci, and loss of function mutations have been identified in the KRIT1/CCM1, MGC4607/CCM2, and PDCD10/CCM3 genes. Interestingly, recent in vivoexperiments shed some light on the roles of CCM1 and CCM2 in cardiovascular development. Inactivation of CCM1leads to early death of murine homozygous embryos at midgestation (Whitehead et al., 2004).
An arterial defect that includes both dilation and constriction of the aorta and some of its branches, a failure in the recruitment of arterial vascular smooth muscle cells, and an abnormal differentiation of the arteries were detected in those embryos, which strongly suggests that CCM1 is involved in arterial morphogenesis and differentiation during early embryonic life. Inactivation of CCM1 and CCM2 in zebraï¬sh also leads to an early death of the embryo with massive dilation of the heart (Mably et al., 2006). This massive enlargement shows the inability of the endocardium to instruct the myocardial muscle to thicken.
Genetics for Klippel-Feil
Mutations in the GDF6 and GDF3 genes have been identified to cause the disease, although some people with the syndrome do not have identified mutations in the GDF6 or GDF3 genes. In this case, the cause of the condition in these individuals is unknown. GDF6 and GDF3 genes provide the body with instructions for making proteins involved in regulating the growth and maturation of bone and cartilage. These proteins actively regulate cell growth in embryonic and adult tissue. GDF6 specifically is involved in the formation of vertebral bones, among others, and establishing boundaries between bones in skeletal development while GDF3 is involved with bone and cartilage growth. Mutations cause reductions in these functional proteins but, it is unclear exactly how a shortage in these proteins leads to incomplete separation of the vertebrae in people with the syndrome. However, when the GDF6 gene was knocked out in mice, the result was the fusion of bones. Only by identifying the link between the genetic etiology and the phenotypic pathoanatomy of Klippel—Feil syndrome will we be able to rationalize the heterogeneity of the syndrome (Tassabehji et al., 2008)
Natural History of CCMs
According to several studies, CCMs are present in about 0.5% of the general population, and they account for 8-15% of all brain and spinal vascular malformations. The prevalence of cavernous malformations in children is estimated to be between 0.37% and 0.53% (up to 1 in 200 children have a CCM, whether symptomatic or asymptomatic). Approximately 25% of all diagnosed cavernous malformations are found in children, with peak ages of incidence 0-2 years (26.8% of children with CCMs) and 13-16 (35.7%). The natural history of pediatric CCHs seems to be more aggressive than in adult patients; these lesions have higher rates of growth and hemorrhage, larger dimensions and often atypical radiological pictures at diagnosis. Other malformations can be present in combination with CCMs, such as small areas where the capillaries or veins do not form correctly (capillary telangiectasias or venous malformations), and this association occurs 8-44% of the time. (Kidwell and Wintermark, 2008).
A study by Zabramski et al (1994) demonstrated that the familial form of cavernous malformations is a dynamic disease; serial MR images revealed changes in the number, size, and imaging characteristics of lesions consistent with acute or resolving hemorrhage. It supported the de novo development of new lesions that had not been previously reported. These findings suggest that patients with familial cavernous malformations require careful follow-up monitoring, and that significant changes in the neurological symptoms warrant repeat MR imaging. It also concludes that surgery should be considered only for lesions that produce repetitive or progressive symptoms. Also, in a study of microsurgical resection of brainstem cavernomas (Ferroli et al., 2005) surgical resection is recommended for superficial lesions and for lesions that can be reached through the anterolateral pontine surface. On the other hand, rebleeding is not more common among patients who first present with bleeding, and it often has little effect on the neurological status of patients and significant morbidity attributable to a brainstem cavernoma occurred in 8% of patients during follow-up monitoring of medium duration (Kupersmith et al., 2001).
Limitations
In our case, the young patient’s relocation affected follow-up. So, the question remains whether the character of the cavernomas have changed or there are new ones. A good clinical impression does not outweight the lack of radiological image because a change in size, number or event may well be asymptotic. Recurrence of an event in the future or/and alteration of the neurological status should shift its management to a more aggressive one. Also, because the patient is a young female, caution should be taken both in the use of oral contraceptives and the type of labor chosen, in case of pregnancy.
Polycystic ovarian syndrome (PCOS) is a syndrome of variable combinations of menstrual irregularity, hirsutism or acne. It can be diagnosed in adolescence. It is a metabolic syndrome characterized by hyperandrogenism and chronic anovulation, and it is among the most common endocrine disorders affecting women of reproductive age. Abnormal glucose tolerance (impaired glucose tolerance (IGT) and diabetes mellitus (DM)), at least in part due to insulin resistance, is present among approximately 30—40% of the affected individuals and is independent of body weight. The exact etiopathophysiology is unclear. The abnormal function of the hypothalamic-pituitary-ovarian axis and the inappropriate gonadotropin secretion are more likely a result rather than a cause of PCOS. The source of androgens may be from the ovaries, adrenals, or both. There is some evidence suggesting that patients have a functional abnormality of cytochrome P450c17, the 17-hydroxylase, which is the rate-limiting enzyme in androgen biosynthesis. (Chau et al., 2009).
Nephrolithiasis is relatively uncommon in the pediatric population. However, it can be a harbinger for systemic metabolic disorder or underlying anatomical abnormalities. Nephrolithiasis in children is considered to be predominantly the result of metabolic rather than infectious causes. Although infections are commonly associated with kidney stones it is unlikely to be the etiological factor. Metabolic predispositions for nephrolithiasis are revealed in more than 40% of the affected children. (Eisenmann, 2007). In our case, nephrolithiasis, could be considered to belong to the cluster of conditions that accompany Klippel-Feil syndrome. The PCOS of our patient does not seem to be related with the other conditions. Glucose intolerance, also relates to the PCOS syndrome than the rest of morbidities of this patient.
Conclusion
It is evident that there is a great combination of CMs which implicates defects during a specific period of embryogenesis. However, this is not an absolute principle; in certain mal-developments of the brain, which, most probably, had originated during the embryonic period, all the other organs retain their normal developmental path. One can only assume that the brain is more vulnerable than any other organ to prenatal as well as natal influences, even if, so far, the possible connection between genetic defects, metabolic disorders and exogenous effects that lead to combined CMs can only be inferred.
We presented a novel combination of extensive cerebral cavernous hemangioma of the brainstem in an adolescent Caucasian female patient, with multiple cavernomatosis, coexistent with Klippel Feil type II, nephrolithiasis, PCOS and glucose intolerance. This is a novel combination that has never been previously reported in the literature.
References
Klippel, M. & Feil, A. (1912). ‘Un Cas D’ Absence des Vertebres Cervicales. Avec Cage Thoracique Remontant Jusqu’a La Base Du Crane (Cage Thoracique Cervicale),’ Nouv Iconog Salpetriere, 25, 223-250. Google Scholar