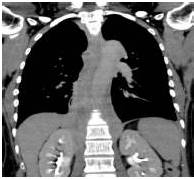
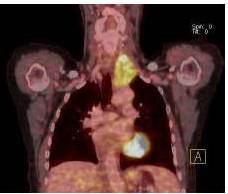
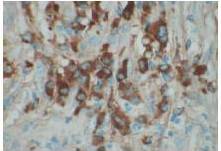
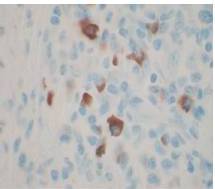
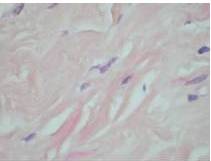
Therefore, the high levels of IgG4+ plasma cells at the time of the recurrence, the number of plasma cells expressing IgG4 > 10, the absence of neoplastic processes, such as carcinoma, lymphoma, or sarcoma, the absence of TPO and Tg antibodies, the breaking of the thyroid capsule, the regression of the tumor, and the decrease in the levels of circulating IgG4s under corticosteroids enabled us to establish the diagnosis of IgG4-related syndrome of the thyroid. Moreover, the (18)FDG-PET/CT did not reveal any other organ affection due to the disease. After six months of treatment with methylprednisolone, with a dose reduction over time, the disease did not progress anymore and IgG4 levels remained within the normal range. At six months, the ultrasound showed a significant decrease of the carotid-jugular mass.
The contribution of IgG4 levels in the diagnosis of IgG4-SD, which shows a sensitivity of 71% to 93% for AIP, is highly debated in the literature. A recent study showed that a threshold of IgG4 levels at 140 mg/dL had a sensitivity of 76%, a specificity of 93%, and positive predictive value of 36%. These rates were 53%, 99%, and 75% with a double threshold of 280 mg/dL for AIP (Lamia, 2011). To date, no data is available for the thyroid, but IgG4 levels of 333 mg/dL in our patient enabled us to confirm the diagnosis. However, IgG4 levels should be cautiously interpreted, depending on the clinical, histopathological, and immunohistochemical context and current or previous treatments administered. Carbonic anhydrase (CA) and lactoferrin antibodies can be found in most patients with AIP (16), antinuclear antibodies (ANAs) can be found in 43% to 75% of cases, and rheumatoid factor (RF) can be found in 13% to 30% of cases (Lamia, 2011). Here, antibodies to CA, RF, and ANA could not be found.
In the present study, we also confirm that (18)FDG-PET/CT provides valuable data for the monitoring of IgG4-SD. It constitutes a test of choice to assess the extent and metabolism of IgG4-SD (Nguyen, 2011). In addition, although we used tamoxifen for its antifibrotic effects, monotherapy with tamoxifen for more than 12 months did not allow for the remission of IgG4-SD, unlike the case in the study by Chacko et al. (Chacko, 2009).
In conclusion, significant similarities exist between RT and IgG4-SD, such as the presence of invasive fibrosis, lymphoplasmacytic infiltration, and good response to steroid therapy, which have been often reported and are well illustrated in the present case. RT associated with a high number of IgG4+ plasma cells, with or without elevated serum IgG4 levels, may constitute the first symptoms of a case of IgG4-SD. The assessment of a patient with thyroid fibrosis should ideally include an immunohistochemical evaluation of plasma cells (IgG+ and IgG4+ cells) and of circulating levels of IgG4s. Treatment with corticosteroids seems to be the cornerstone. It favorably influences the course of the disease. The monitoring of circulating IgG4s in the patient under treatment with corticosteroids correlated with a regression of the tumor in the present case. In addition, the recurrence of the disease in the patient under tamoxifen monotherapy is challenging. Therefore, more data is needed to clarify the link or the difference between these two entities.
References
Chacko, S., et al. (2009). “Treatment of Hyper-IgG4 Disease with Sequential Corticosteroids and Tamoxifen – Case Report and Review of the Literature,” Clinical Nephrology, 72:414-7.
Publisher – Google Scholar
Dahlgren, M., Khosroshahi, A., Nielsen, G. P., Deshpande, V. & Stone, J. H. (2010). “Riedel’s Thyroiditis and Multifocal Fibrosclerosis are Part of the IgG4-Related Systemic Disease Spectrum,” Arthritis Care & Research (Hoboken), 62:1312-8.
Publisher – Google Scholar
Ebbo, M., Grados, A., Daniel, L., Vély, F., Harlé, J. R., Pavic, M. & Schleinitz, N. (2012). “IgG4-Related Systemic Disease: Emergence of a New Systemic Disease? Literature Review,” La Revue de Medecine Interne, 33:23-34.
Publisher – Google Scholar
Erdoğan, M. F., Anil, C., Türkçapar, N., Ozkaramanli, D., Sak, S. D. & Erdoğan, G. ( 2009). “A Case of Riedel’s Thyroiditis with Pleural and Pericardial Effusions,” Endocrine, 35:297-301.
Publisher – Google Scholar
Fatourechi, M. M., Hay, I. D., McIver, B., Sebo, T. J. & Fatourechi, V. (2011). “Invasive Fibrous Thyroiditis (Riedel Thyroiditis): The Mayo Clinic Experience, 1976-2008,” Thyroid, 21:765-72.
Publisher – Google Scholar
Hamano, H., Kawa, S., Horiuchi, A., Unno, H., Furuya, N., et al. (2001). “High Serum IgG4 Concentrations in Patients with Sclerosing Pancreatitis,” The New England Journal of Medicine, 344:732-8.
Publisher – Google Scholar
Hennessey, J. V. (2011). “Clinical Review: Riedel’s Thyroiditis: A Clinical Review,” The Journal of Clinical Endocrinology & Metabolism, 96:3031-41.
Publisher – Google Scholar
Kakudo, K., Li, Y., Taniguchi, E., Mori, I., Ozaki, T., Nishihara, E., Matsuzuka, F. & Miyauchi, A. (2012). “IgG4-Related Disease of the Thyroid Glands,” Endocrine Journal, 59:273-81.
Publisher – Google Scholar
Kallel, L., Naija, N., Boubaker, J. & Filali, A. (2011). “La Pancréatite Auto Immune: Revue Systématique de la littérature,”La tunisie Medicale, 89:221-30.
Publisher – Google Scholar
Michels, J.- J. & Blanc-Fournier, C. (2008). “What Diagnosis Can be Made by the Pathologist in a Case of Sclerosing Thyroiditis?,” Annales de Pathologie, 28:263-7.
Publisher – Google Scholar
Nguyen, V. X., De Petris, G. & Nguyen, B. D. (2011). “Usefulness of PET/CT Imaging in Systemic IgG4-Related Sclerosing Disease. A Report of Three Cases,” JOP. Journal of the Pancreas, 12:297-305.
Publisher – Google Scholar
Oriot, P., Malvaux, P., Waignein, F., Delcourt, A., Doyen, C., Rousseau, E., Baudry, G. & Dechambre, S. (2012). “Nodular Sclerosing Hodgkin’s Disease Mimicking Riedel’s Invasive Fibrous Thyroiditis,” Annales d’Endocrinologie (Paris),73(5) 492-6.
Publisher – Google Scholar
Papi, G. & LiVolsi, V. A. (2004). “Current Concepts on Riedel Thyroiditis,” American Journal of Clinical Pathology, 121:50-63.
Publisher – Google Scholar
Perimenis, P., Marcelli, S., Leteurtre, E., Vantyghem, M. C. & Wémeau, J. L. (2008). “Riedel’s Thyroiditis: Current Aspects,” Presse Medicale, 37:1015-21.
Publisher – Google Scholar
Pusztaszeri, M., et al .(2012). “Riedel’s Thyroiditis with Increased IgG4 Plasma Cells: Evidence for an Underlying IgG4 Related Sclerosing Disease?,” Thyroid, 22 (9):964-8.
Publisher – Google Scholar
Stone, J. H., Zen, Y. & Deshpande, V. (2012). “IgG4-Related Disease,” The New England Journal of Medicine, 366:539-551.
Publisher – Google Scholar
Vaidya, B., Harris, P. E., Barrett, P. & Kendall-Taylor, P. (1997). “Corticosteroid Therapy in Riedel’s Thyroiditis,”Postgraduate Medical Journal, 73:817-9.
Publisher – Google Scholar
Yasmeen, T., Khan, S., Patel, S. G., Reeves, W. A., Gonsch, F. A., de Bustros, A. & Kaplan, E. L. (2002). “Clinical Case Seminar: Riedel’s Thyroiditis: Report of a Case Complicated by Spontaneous Hypoparathyroidism, Recurrent Laryngeal Nerve Injury, and Horner’s Syndrome,” The Journal of Clinical Endocrinology & Metabolism, 87:3543-7.
Publisher – Google Scholar