Muhammed Mustafa Demirpence, Mithat Bahceci, Devrim Dolek, Fusun Salgur, Ahmet Gorgel and Pelin Tutuncuoglu
Izmir Ataturk Training and Research Hospital, Department of Endocrinology
Volume 2013 (2013),
Article ID 511990,
International Journal of Case Reports in Medicine,
6 pages,
DOI: 10.5171/2013.511990
Received date: 25 January 2013; Accepted date: 25 February 2013; Published date: 27 April 2013
Academic Editor: Ewa Bar-Andziak
Cite this Article as:
Muhammed Mustafa Demirpence, Mithat Bahceci, Devrim Dolek, Fusun Salgur, Ahmet Gorgel and Pelin Tutuncuoglu (2013), "A Very Rare Assocıatıon; Coexıstence of Breast Cancer, Pheochromocytoma and Neurofıbromatosıs Type 1 in a Female Patıent" International Journal of Case Reports in Medicine, Vol. 2013 (2013), Article ID 511990, DOI: 10.5171/2013.511990
Neurofibromatosis type 1 (NF1) is an autosomal dominant condition and patients with NF-1 risk malignant diseases, such as breast cancer. We aim to report a coexistence of both breast cancer and phochromocytoma in a NF1 patient. A female patient (47yr) with adrenal incidentaloma who had undergone? modified mastectomy for invasive ductal and lobular carcinoma 5 years ago. NF1 was diagnosed with multiple cafe au lait spots, discrete cutaneous neurofibromas over the patient’s body and freckling in the axillary region. Patient’s father who died at the age of 48 due to pancreatic cancer with liver metastasis and son (13 yr) had similar cafe au lait spots. She was still hypertensive (180/105 mmHg) under the triple anti-hypertensive drugs. Abdominal CT scan showed a 9 × 10 cm diameter mass arising from left adrenal gland with mixed consistency. Urinary metanephrine and normetanephrine levels (24 hourly) were extremely high (11125 and 15042 mic.g, respectively). We diagnosed pheochromocytoma with these results. Multiple endocrine neoplasia type 2 could not be detected and patient was redirected to a surgery clinic under suitable conditions. Although malignant tumors are reported four times as often in NF-1 patients than in the general population, coexistence of breast cancer and pheochromocytoma in NF-1 patients have never been reported before. Here we report probably the first coexistence of NF1, breast cancer and pheochromocytoma.
Keywords: Neurofibromatosis type 1, breast cancer, pheochromocytoma.
Introduction
Neurofibromatosis type 1 (NF1, OMIM 162200), formerly known as von Recklinghausen disease, is an autosomal dominant condition affecting one in 3000 live births [In a research study by Huson (1989)]. It is mainly caused by mutation of the NF-1 gene, which is located in chromosome band 17q11.2 [Nemoto et al(2006) mentioned]. Diagnosis is confirmed in the presence of 2 or more of the features listed: six or more café au lait macules (larger than 5 mm in greatest diameter in prepubertal individuals, larger than 15 mm in greatest diameter in postpubertal individuals); freckling in the axillary or inguinal regions; optic pathway glioma; 2 or more Lisch nodules; a distinctive osseous lesion, such as sphenoid dysplasia or thinning of the long bone cortex, with or without pseudoarthrosis; a first-degree relative with NF-1 according to the above criteria, 2 or more neurofibromas of any type or 1 plexiform neurofibroma [İn the book by Hottinger and Khakoo (2007) ]. Complete clinical manifestations at 8-10 years of age. Symptoms and severity of the disorder may vary among members of an affected family.
The NF1 gene is a tumor suppressor gene and it encodes neurofibromin, a cytoplasmic protein that is expressed in neurons, schwann cells, oligodendrocytes, astrocytes and leukocytes. Neurofibromin is a negative regulator of the Rasoncogene, the inactivation of which leads to cell proliferation and tumor development. For that reason, patients with NF-1 have about 3-15% additional risk of malignant disease in their lifetime and these patients are at a fourfold increased risk of cancer compared with the general population [İn two research studies by Airewele (2001) and Sorensen(1986)]. Malignant tumors can arise in the nervous and non-nervous system and patients have a greatly increased relative risk of developing gliomas, malignant peripheral nerve sheath tumors, juvenile chronic myelomonocytic leukaemia, rhabdomyosarcoma and pheochromocytoma. Walker L et al. reported (2006) that the overall risk of cancer was 2.7 times higher in this cohort of NF1 patients than in the general population. In this study, the most frequent types of cancer are connective tissue and brain tumors, and there is no statistically significant excess of cancers at other sites. On the other hand, Sharif S. et al. [2007] showed that breast cancer should be considered a common association in NF1, and that affected women will require screening for this from the age of 40. Although there is a myriad of case reports about the association of NF1 and breast cancer or pheochromocytoma separately, we could not find any coexitence of both breast cancer and pheochromocytoma in patient with NF1 in medical literature (www.pubmed.com). Therefore, this is probably the first NF1 case who had breast cancer and pheochromocytoma.
Case
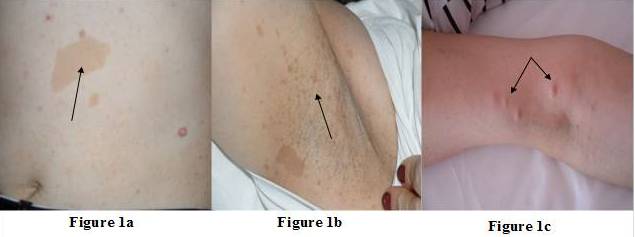
A female patient (47yr) was admitted to our clinic due to adrenal incidentaloma. She had undergone right modified mastectomy for the treatment of invasive ductal and lobular carcinoma (T2N0M0) of breast 5 years ago. In the physical examination, we noticed multiple cafe au lait spots, freckling in the axillary region and discrete cutaneous neurofibromas over the patient’s body (Figure 1a, b and c) and NF1 was diagnosed. Hypertension was diagnosed 1 year ago (BP: 180/105 mmHg) and she is currently requiring three antihypertensive drugs (ramipril and hydrochlorothiazide; 5/12.5 mg and nifedipine 10 mg). Patient’s father died at the age of 48 due to pancreatic cancer with liver metastasis and son (13 yr) had similar cafe au lait spots.
[Figure 1a, B and C]: Multiple Cafe au Lait Spots, Freckling in the Axillary Region and Discrete Cutaneous Neurofibromas over the Patient’s Body
Laboratory and Imaging Examinations
Abdominal ultrasonographic evaluation revealed a mass lesion in left adrenal gland and afterwards abdominal CT scan showed a 9 × 10 cm diameter mass arising from left adrenal gland with mixed density (Figure 2). Neither lymphadenopathy nor distant metastasis was detected. In the clinical evaluation of adrenal incidentaloma, we detected very high 24 hour urinary metanephrines and normetanephrines levels (11125 and 15042 microgram, respectively). Dynamic tests for Cushing’s syndrome and primary hyperaldosteronism were normal (Serum cortisol level after 1 mgdexametasone: 1,55 ug/dL, serum aldosterone level: 356 pg/mL). With these findings we diagnosed pheochromocytoma, and left adrenalectomy and tumor biopsy were performed. In the histopathological examination, adrenal tumor had a potential for a biologically aggressive behavior [total score was 6 according to pheochromocytoma of the adrenal gland scaled score (PASS)][ [In the research study by Thompson (2002)]]. Cutaneous neurofibroma was diagnosed in skin biopsy. Patient was also screeneed for multiple endocrine neoplasia type 2, but the results were negative (Serum parathyroid hormone: 41 pg/mL, calcitonin: 1 pg/mL).
[Figure 2]: Abdominal CT Scan Showed a 9 × 10 Cm Diameter Mass Arising from Left Adrenal Gland with Mixed Density
Discussion
In our patient presenting diagnostic criteria of NF1 (Six or more café au lait macules, larger than 15 mm in greatest diameter, freckling in the axillary region and 2 or more neurofibromas) we diagnosed NF1. As mentioned above, NF1 is mainly caused by mutation of the NF-1 gene [Nemoto et al (2006) mentioned]. This gene is a tumour suppressor gene and it encodes neurofibromin. Inactivation of neurofibromin leads to cell proliferation and tumor development [İn two research studies by Airewele (2001) and Sorensen(1986)]. In addition to NF1, germline mutations causing a predisposition to the development of malignancy in neurofibromatosis, somatic mutations of NF1 have been associated with a number of sporadic malignancies such as breast cancer, and others [In a research study by Teschendorff (2007)].
Our patient had typical findings of pheochromocytoma. She was hypertensive (180/105 mmHg), and she had a left adrenal mass and very high 24 hour urinary metanephrine and normetanephrine levels. The incidence of pheochromocytoma in association with NF1 has been reported to be 0.1% to 5.7%, but autopsy studies found the prevalence to be up to 13% [Walther et al (1999) mentioned ]. Pheochromocytomas are most commonly diagnosed in adulthood and are rare in children with NF1 [Walther et al (1999) mentioned]. In fact, pheochromocytoma appeared in adulthood (47 yr) in our patient and this finding also support that pheochromocytoma is most commonly diagnosed in adulthood period in patients with NF1.
Coexistence of pheochromocytoma and NF1 is well known but the most extraordinary finding of our patient is the coexistence of both breast cancer and pheochomocytoma in the same patient. This is probably the first case of NF1 ; because we screened the medical literature with the key words of “neurofibromatosis type 1, breast cancer and pheochromocytoma” by Pubmed (www.pubmed.com), but we could not find any report of coexistence of all three (NF1, breast cancer and pheochromocytoma). Therefore, this is likely the first case of NF1 presenting with both breast cancer and pheochromocytoma.
Tumors in patients with NF1 may also have a different natural history from those occurring sporadically, and require a specific approach to their detection and management. The NF1 gene has a very high rate of spontaneous mutations, resulting in 50% of cases arising de novo [In a research study by Littler (1990)]. Mutation of NF1 gene in our patient may be different and de novo; and such mutation may have greater probability of associating with malignant disease than other known mutations. The adrenal tumor of our patient had a potential for biologically aggressive behavior [total PASS score is 6 [In the research study by Thompson (2002)], whereas pheochromocytomas in NF—1 were similar to sporadic pheochromocytoma in terms of malignance potential. The main deficiency of our study is that genetic analysis has not been realized. We could not determine the type of NF1 gene mutation of our patient due to financial reasons; the cost of this genetic analysis is too expensive and the social security foundation of our country does not meet such analysis.
Conclusions
1-Women with NF1 have risk of developing breast cancer and are advised to have yearly screening for breast cancer from the age of 40. 2-Although NF1 patients may have risk of breast cancer and pheochromocytoma separately, coexistence of both diseases in the same patient is very rare and, probably, this is the first case report to include a female NF1 patient who had both breast cancer and pheochromocytoma.