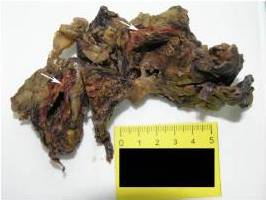
The patient developed severe anemia, bilateral lung edema and hydrothorax along with hemorrhage from the postoperative wound. He was treated with supportive therapy and blood transfusions but with no response to therapy. He died 19 days after nephrectomy that was three and a half months after primary diagnosis of the adrenal tumour.
Literature Review, Discussion and Conclusion
In the studies performed by Meis-Kindblom and Kindblom (1998) and Deyrup et al. (2009), it was shown that except in the skin, hemangiosarcoma can occur in the mediastinum, breast, mesentery, deep soft tissue of upper extremity and visceral organs, usually liver. They usually present as multinodular hemorrhagic masses that range in size from a few millimeters to several centimeters in diameter. Extensive hemorrhage is a characteristic feature of most tumours and it can mimic chronic hematoma. Although most cases are sporadic, important predisposing conditions include chronic lymphedema, exposure to toxins (e.g. vinyl chloride), and foreign bodies (e.g. arteriovenous fistulas). Association with radiation therapy was described in female patients treated for breast carcinoma (Yap et al., 2002).
According to Rosen et al. (1988), Meis-Kindblom and Kindblom (1998) and Deyrup et al. (2009), most hemangiosarcomas have a poor outcome, regardless of grade. After surgery, local recurrences develop in about one fifth of patients and one half may be expected to die within the first year after diagnosis. Older age, retroperitoneal location, large size and high Ki-67 values correlate with poor outcome. Systemic chemotherapy is usually administered in locally recurrent or metastatic disease or with less invasive surgical approach in young patients. No optimal adjuvant chemotherapy has yet been identified.
PAA is extremely rare. To our knowledge there are 20 cases of PAA described to date in the English literature (Kareti et al., 1988; Livatidou et al., 1991; Bosco et al., 1991; Wenig et al., 1994; Otal et al., 1999; Ben Izhak et al., 1999; Krüger et al., 2001; Croitoru et al., 2001;Invitti et al., 2001; Pasqual et al., 2002;Lepoutre-Lussey et al., 2012; Derlin et al., 2012).
The first case of PAA was reported by Kareti et al. in 1988. In their case, a 54-year-old man was found to have left adrenal mass on abdominal CT scan. The tumour was surgically removed. Diagnosis of adrenal hemangiosarcoma, supported by findings of immunoperoxidase and ultrastructural studies, was made.
The largest series with 9 cases of PAA was described by Wenig et al. in 1994. In their study, patient age range was 45-85 years with most occurring in sixth and seventh decades of life with no sex predilection. The size of the tumour ranged from 6 to 10 cm in greatest dimension. Mostly it presented as an isolated adrenal mass either asymptomatic or causing non-specific symptoms like pain in the adrenal area occupied by the tumour mass as in our case, or in patients presented with slight fever, anorexia and fatigue. Three patients were alive in the time of study, three died from lung metastases and 3 others died from unrelated causes.
The youngest patient diagnosed with PAA was a 34-year old that had overt Cushing’s syndrome due to ACTH-secreting pituitary adenoma and PAA described by Invitti et al. in 2001. Some patients can have symptoms similar to paraneoplastic syndrome as was described by Bosco et al. (1994).
Although most described cases of PAA appear sporadically, chronic arsenical intoxication (especially of vineyard cultivators) may be an important causative factor in the pathogenesis of the disease as was suggested by Livaditou et al. (1991).
According to Otal et al. (1999), the radiology workup may suggest only an indistinct malignancy, since radiological features of hemangiosarcoma are nonspecific.
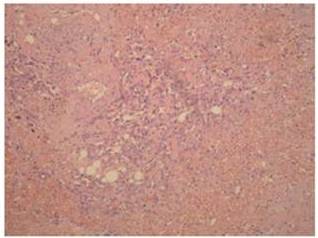
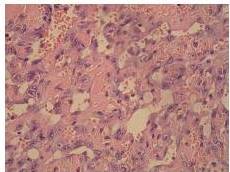
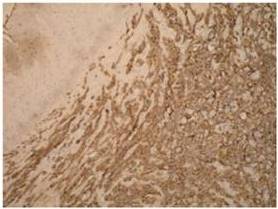
Definitive diagnosis of PAA, as for hemangiosarcomas that arise in other sites is based on histology and immunohistochemistry. According to Meis-Kindblom and Kindblom (1998), light microscopy findings of various atypical cells forming rudimentary vascular channels should raise suspicion for hemangiosarcoma. Immunohistochemically hemangiosarcomas express CD31 and CD34, the fact that can be helpful in confirming the diagnosis.
PAA is highly infiltrative beyond its clinically apparent borders and is often multifocal. Local control has a high failure rate. Patients usually die of extensive haemorrhage or tissue infarctions as was described by Krüger et al. (2001). In their case a 70-year-old man with PAA died 3 weeks after tumour resection due to intestinal infarction and acute renal failure. There were no distant metastases.
As for hemangiosarcomas arising in other sites, initially extensive surgical procedure with removal of whole periadrenal tissue and locoregional lymph nodes is considered to be a treatment of choice for PAA as was argued by Pasqual et al. (2002).
More extensive initial surgery was considered in a case reported by Lepoutre-Lussey et al. in 2012. They presented a 35 year-old man with combined PAA and functioning adrenocortical adenoma. The right adrenalectomy was performed with removal of periadrenal fat tissue. After histological diagnosis, the patient was suggested for another more radical operation with ipsilateral nephrectomy and psoas removal along with omentectomy. Because there was no residual disease and the young age of the patient, a conservative strategy was preferred and the patient received 4 cycles of chemotherapy (adriamycin/ifosfamide). After 2 years he had no signs of relapse.
Chugh et al. (2009) performed the phase II trial study in which they assessed Imatinib efficacy in treating 10 different subtypes of advanced sarcomas. Among different sarcoma types there were 16 patients with stage IV (proven metastatic or locally advanced disease) hemangiosarcomas. Imatinib did not prove as an efficient drug in advanced hemangiosarcoma. In our case the patient developed PAA while treated with Imatinib for CML, so indirectly our case support findings from the described trial.
From the information available at the website eHealthMe (accessed 13 February 2013), 19753 people have been reported to have side effects when taking Gleevec (Imatinib). Among them, 2 women (0, 01%), older then 60 had hemangiosarcoma. They were taking Gleevec between 6-12 months in the time hemangiosarcoma was diagnosed. We cannot say if there is a correlation between occurrence of hemangiosarcoma and Gleevec therapy in our patient.
Our patient was first treated with adrenalectomy and removal of periadrenal fat tissue, but eventually developed complications and underwent ipsilateral nephrectomy and omentectomy. A review of the literature underlines the poor clinical outcome of PAA with most patients dying because of hemorrhage and recurrence of disease as in our case.
Radiological findings are nonspecific so the diagnosis of PAA can be made only microscopically. Because of the small number of cases described in the literature, the question remains whether relaparotomy and more radical en bloc surgical procedure should be done after histological diagnosis of PAA or some other type of treatment should be applied.
References
Ben-Izhak, O., Auslander, L., Rabinson, S., Lichtig, C. & Sternberg, A. (1992). “Epithelioid Hemangiosarcoma of the Adrenal Gland with Cytokeratin Expression. Report of a Case with Accompanying Mesenteric Fibromatosis,” The Cancer Journal, 69 (7) 1808-1812.
Publisher – Google Scholar
Bosco, P. J., Silverman, M. L. & Zinman, L. M. (1991). ‘Primary Hemangiosarcoma of Adrenal Gland Presenting as Paraneoplastic Syndrome: Case Report,’ Journal of Urology, 146 (4) 1101-1103.
Chugh, R., Wathen, J. K., Maki, R. G., Benjamin, R. S., Shreyaskumar, R. P., Myers, P. A., Priebat, D. A., Reinke, D. K., Thomas, D. G., Keohan, M. L., Samuels, B. L. & Baker, L. H. (2009). “Phase II Multicenter Trial of Imatinib in 10 Histologic Subtypes of Sarcoma Using a Bayesian Hierarchical Statistical Model,” Journal of Clinical Oncology, 27 (19) 3148-3153.
Publisher – Google Scholar
Croitoru, A. G., Klausner, A. P., McWilliams, G. & Unger, P. D. (2001). “Primary Epithelioid Hemangiosarcoma of the Adrenal Gland,” Annals of Diagnostic Pathology, 5 (5) 300-303.
Publisher – Google Scholar – British Library Direct
Derlin, T., Clauditz, T. S. & Habermann, C. R. (2012). “Adrenal Epithelioid Hemangiosarcoma Metastatic to the Epicardium: Diagnosis by 18F-FDG PET/CT,” Clinical Nuclear Medicine, 37 (9) 914-5.
Publisher – Google Scholar
Deyrup, A. T., Miettinen, M., North, P. E., Khoury J. D., Tighiouart, M., Spunt, S. L., Parham, D., Weiss, S. W. & Shebata, B. M. (2009). “Hemangiosarcomas Arising in the Viscera and Soft Tissue of Children and Young Adults: A Clinicopathologic Study of 15 Cases,” The American Journal of Surgical Pathology, 33 (2) 264-9.
Publisher – Google Scholar
Invitti, C., Pecori, G. F., Cavagnini, F. & Sonzogni, A. (2001). “Unusual Association of Adrenal Hemangiosarcoma and Cushing’s Disease,” Hormone Research, 56 (3-4) 124-129.
Publisher – Google Scholar – British Library Direct
Kareti, L. R., Katlein, S., Siew, S. & Blauvelt, A. (1988). ‘Hemangiosarcoma of the Adrenal Gland,’ Archives of Pathology & Laboratory Medicine, 112 (11) 1163-1165.
Krüger, S., Kujath, P., Johannisson, R. & Feller, A. C. (2001). ‘Primary Epithelioid Hemangiosarcoma of the Adrenal Gland – Case Report and Review of the Literature,’ Tumori , 87 (4) 262-265.
Lepoutre-Lussey, C., Rousseau, A., Al Ghuzlan, A., Amar, L., Hignette, C., Cioffi, A., Zinzindohoue, F., Leboulleux, S. & Plouin, P. F. (2012). “Primary Adrenal Hemangiosarcoma and Functioning Adrenocortical Adenoma: An Exceptional Combined Tumor,” European Journal of Endocrinology, 166 (1) 131-135.
Publisher – Google Scholar
Livatidou, A., Alexiou, G., Floros, D., Filippidis, T., Dosios, T. & Bays, D. (1991). “Epithelioid Hemangiosarcoma of the Adrenal Gland Associated with Chronic Arsenical Intoxication?,” Pathology, Research and Practice, 187 (2-3) 284-9.
Publisher – Google Scholar
Meis-Kindblom, J. M. & Kindblom, L.- G. (1998). “Hemangiosarcoma of Soft Tissue: A Study of 80 Cases,” The American Journal of Surgical Pathology, 22 (6) 683-697.
Publisher – Google Scholar – British Library Direct
Otal, P., Escourrou, G., Mazerolles, C., D’Othee, B. J., Mezgahni, S., Nusso, S., Colombier, D., Rousseau, H. &
Joffre, F. (1999). “Imaging Features of Uncommon Adrenal Masses with Histopathologic Correlation,” Radiographics, 19 (3) 569-581.
Publisher – Google Scholar – British Library Direct
Pasqual, E., Bertolissi, F., Grimaldi, F., Beltrami, C. A., Scott, C. A., Bachetti, S., Waclaw, B. U. & Cagoli, P. P. (2002). “Adrenal Hemangiosarcoma: Report of a Case,” Surgery today, 32 (6) 563-565.
Publisher – Google Scholar – British Library Direct
Rosen, P. P., Kimmel, M. & Ernsberger, D. (1988). “Mammary Hemangiosarcoma: The Prognostic Significance of Tumor Differentiation,” The Cancer Journal, 62 (10) 2145—2151.
Publisher – Google Scholar
Wenig, B. M., Abbondanzo, S. L. & Heffes, C. S. (1994). “Epithelioid Hemangiosarcoma of the Adrenal Glands. A Clinicopathologic Study of Nine Cases with a Discussion of the Implications of Finding “Epithelial-Specific” Markers,” The American Journal of Surgical Pathology, 18 (1) 62-73.
Publisher – Google Scholar – British Library Direct
Yap, J., Chuba, P. J., Thomas, R., Aref, A., Lucas, D., Severson, R. K. & Hamre, M. (2002). “Sarcoma as a Second Malignancy after Treatment for Breast Cancer,” International Journal of Radiation Oncology* Biology* Physics, 52 (5) 1231—1237.
Publisher – Google Scholar – British Library Direct
(Retrieved February, 2013), //http://www.ehealthme.com/ds/gleevec/hemangiosarcoma
Publisher
Ethical Issues:
The authors claim that the anonymity of the patient is protected.
The authors have no competing interests.