Lipoprotein-Lipase Deficiency: Treatment with Omega-3 Fatty Acids and Colestyramine for Severe Gestational Hypertriglyceridaemia and Follow up over Twenty Years
Charles van Heyningen, Irina Cozma and Malani Arasaradnam
Clinical Laboratories, University Hospital Aintree, Lower Lane, Liverpool, England, UK
Volume 2013 (2013),
Article ID 810803,
International Journal of Case Reports in Medicine,
7 pages,
DOI: 10.5171/2013.810803
Received date: 26 January 2013; Accepted date: 17 February 2013; Published date: 31 May 2013
Academic Editor: John D. Brunzell
Cite this Article as:
Charles van Heyningen, Irina Cozma and Malani Arasaradnam (2013), "Lipoprotein-Lipase Deficiency: Treatment with Omega-3 Fatty Acids and Colestyramine for Severe Gestational Hypertriglyceridaemia and Follow up over Twenty Years," International Journal of Case Reports in Medicine, Vol. 2013 (2013),
Article ID 810803, DOI: 10.5171/2013.810803
We describe the case of a Chinese woman with lipoprotein lipase (LPL) deficiency treated with omega-3 fatty acids and colestyramine during pregnancy at the age of 31 years, and her subsequent management with other lipid regulating drugs over the following 21 years. The coexistence of LPL gene and ApoA2 gene mutations, gestational diabetes and ApoE2 heterozygosity resulted in marked hypertriglyceridemia and acute pancreatitis during her fourth pregnancy. Treatment of the severe gestational hypertriglyceridaemia with omega-3 fatty acids was effective during pregnancy before the introduction of colestyramine. Our findings indicate that colestyramine should not be used in pregnancy as it may increase triglyceride levels and cause acute pancreatitis. After this pregnancy she was managed using a combination of lipid regulating drugs. Gene replacement therapy for LPL deficiency provides hope as a future effective therapy for this rare serious inherited condition.
Lipoprotein lipase (LPL) deficiency is a rare inherited disorder of lipoprotein metabolism characterised by severely raised serum triglycerides with an associated high risk for pancreatitis. Treatment with omega-3 fatty acids enhances the clearance of plasma chylomicrons and reduces production of hepatic VLDL. There are very few reports on the use of omega-3 fatty acids in LPL-deficiency and no studies on the use of bile acid sequestrants during pregnancy. We describe the case of a pregnant woman with LPL deficiency treated with omega-3 fatty acids and colestyramine during the pregnancy and her management with other lipid regulating drugs over 21 years. We consider the potential future use of LPL gene therapy which has recently become available in Europe.
Case Report
A 31-year-old Chinese woman was referred to our lipid clinic at 20 weeks gestation (gravida 4, para 3) with severe hypertriglyceridemia. Her plasma had appeared “grossly lipaemic” at 13 weeks gestation when she presented with recurrent abdominal pain. At that time, serum triglyceride and total cholesterol levels were 90 mmol/L and 24 mmol/L respectively. No eruptive xanthomata were seen but she had severe lipaemia retinalis. She weighed 58 kg (BMI 25 kg/sq.m.) which was 8 kg above her non-pregnant weight and her blood pressure was low at 90/70 mmHg. She had been found to have hyperlipidaemia in China at the age of 21 years. Two previous pregnancies had been without complications. Her father had hypertension, hyperlipidaemia, smoked and died from a myocardial infarction at age 56 years. There was no further known family history of cardiovascular disease. She did not drink alcohol or take any medication. Hypothyroidism and renal insufficiency were excluded (normal serum thyroid stimulating hormone and creatinine) but gestational diabetes was diagnosed with an oral glucose tolerance test (2 hour plasma glucose of 9.5 mmol/L after a 75 grams glucose load).
Analysis of serum VLDL apolipoproteins by isoelectric focusing showed that apoC-II was present in normal amounts. Apoprotein E phenotyping showed an E2/E3 variant. During the pregnancy, samples were collected at 21 weeks gestation for LPL activity assay. Blood was collected into lithium heparin tubes before and 20 minutes after an intravenous injection of 1700 units heparin. Plasma triglycerides were 38 mmol/Lbefore and 29 mmol/L after heparin. Post—heparin plasma lipoprotein lipase (LPL) activity, measured by hydrolysis of labelled triolein emulsion after inhibition with specific antisera (Diabetes and Lipid Laboratories, St Bartholomew’s Hospital, London), was markedly reduced at 6.9 nmoles/FFA/min/ml (normal 90-270). These results are compatible with type I hyperlipidaemia due to LPL deficiency.
On genetic analysis the patient has both LPL gene and ApoA5 gene mutations (see results below).
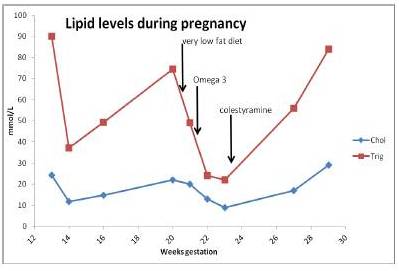
The patient was advised to follow a very low fat diet containing 18-20 g fat, 90 g protein and 1200-1300 kilocalories per day. This reduced her serum triglycerides to 49 mmol/L over 12 days. By taking prescribed omega-3-fatty acid capsules 10 g/day with a low fat diet, her serum triglycerides were further reduced by 50% to 24 mmol/L over 9 days. Following this she was prescribed colestyramine 4 g daily in an ante-natal clinic. Serum triglycerides rose to 56 mmol/L over the next 3 weeks and she developed abdominal pain at 29 weeks gestation. She underwent a Caesarean section because of foetal distress and delivered a normal premature infant. During surgery it was noted that her pancreas was inflamed and that serum amylase was four times the upper limit of normal, confirming a diagnosis of acute pancreatitis. Serum triglycerides were 84 mmol/L after surgery. Changes in serum lipid levels during this pregnancy are shown in figure 1. She made a good recovery and 2 months after delivery her serum triglycerides were down to 5 mmol/L on bezafibrate 600 mg daily.
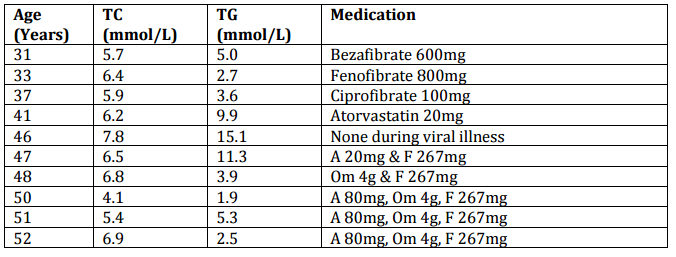
She has been followed up regularly over 21 years following this pregnancy (see table 1 for lipid results and treatment). She developed type 2 diabetes mellitus at age 51 years. On combined therapy with a statin (atorvastatin 80 mg/day), fibrate (fenofibrate 267 mg/day) and fish-oils (omega-3 fatty acids 4 g/day) her total cholesterol is 6.9 mmol/L and triglycerides 2.5 mmol/L. Her diabetes is well controlled on metformin.
Her three children, aged 20, 24 and 27 years, have normal lipid levels. Her brother was found to have serum triglycerides of 25.7 mmol/L at age 40 years and these have been reduced to 2.4 mmol/L on a combination of Atorvastatin 80mg, Niaspan 2 grams, Fenofibrate 267 mg and omega-3 fatty acids 4 g/day.
Genetic Analysis Results
A blood sample was screened by DNA sequencing for changes within the LPL and APOA5 genes, and for large deletions/duplications within the LPL gene. DNA Sequencing identified the following changes:
1. Heterozygous for LPL c.568G>A; p.Glu190Lys (also known as p.E190K)
2. Homozygous for APOA5 c.-3A>G; p. (?)
3. Heterozygous for APOA5 c.553G>T; p.Gly185Cys (also known as p.G185C)
LPL c.568G>A is not considered to be a single nucleotide polymorphism (SNP), and does not appear on dbSNP or the Exome Variant Server database. It has not been reported in the literature. In silico analysis is not conclusive; however it is highly conserved at both the nucleotide and amino acid level, and PolyPhen2 predicts it to be probably damaging. On balance, we consider LPL c.568G>A to most likely be a pathogenic variant. No other point mutations were found within LPL (and no deletions or duplications identified by MLPA)
APOA5 c.-3A>G is a documented SNP (rs651821), occurring at a moderately high frequency with a Minor Allele Freq reported as 7% (NCBI), and 7-14% ( Exome Variant Server). However, it is situated within the Kozak Consensus Sequence, and has been shown in a number of reports in the literature to be associated with hypertriglycerideaemia, and play a role in APOA5 translation in vitro (although a direct role in ApoAV protein levels is less clear). We consider this to be a functional polymorphism which may (particularly in the homozygous state) play a role in plasma triglygeride levels.
APOA5 c.553G>T is a documented SNP (rs2075291), occurring at a generally low frequency (although significantly higher in Asian populations) with a Minor Allele Freq reported as 0.6-6% (NCBI) and 0.06-0.2% (Exome Variant Server). The amino acid is moderately conserved across species, occurring within the receptor binding domain. There is a large physiochemical difference between the normal and substituted residue, and both SIFT and PolyPhen2 predict the change to be damaging. A number of reports in the literature demonstrate an association with hypertriglycerideaemia and a reduction in LPL activation in vitro. We would consider this to most likely be a pathogenic variant.
Discussion
Our patient presented in the second trimester of her fourth pregnancy. She had several coexisting factors that each contributed to an increase in the level of triglycerides in plasma (a LPL gene mutation, two ApoA5 gene mutations, gestational diabetes mellitus and apo-E2 heterozygosity), resulting in a very severe hypertriglyceridemia on presentation.
LPL deficiency is a rare condition. Prevalence for homozygous genetic LPL deficiency varies between 1:1,000,000 and 1:5,000 in French Quebec and in the Netherlands it is estimated to be at approximately 1:500,000 (Nierman et.al. 2005). LPL is a lipolytic enzyme which hydrolyses triglyceride-rich lipoproteins to glycerol and free fatty acids. Deficiency in this enzyme results in high plasma triglyceride concentrations and low HDL, with a consequent increase in the risk of atherosclerosis. The enzyme deficiency may be caused by loss of function mutations of the LPL gene located on chromosome 8. Recurrent abdominal pain is a common presenting feature of this condition (as seen in our patient). The chylomicron accumulation and severe hypertriglyceridaemia may also cause acute pancreatitis.
Heterozygotes for LPL deficiency exhibit a 50% decrease of LPL enzyme activity in plasma following intravenous administration of heparin. Mild lipid abnormalities not associated with familial LPL deficiency have been reported with common variants of the LPL gene. Such variants may be associated with hypertriglyceridemia in the presence of apoE2, diabetes mellitus or familial combined hyperlipidemia.
More than 220 disease-causing mutations of the LPL gene have been identified. At least 28 missense mutations associated with markedly reduced or absent LPL activity have been described.
Unlike the other members of the multigene apolipoprotein family, APOA5 was discovered only recently. The apolipoprotein A5 (apoA5) protein is present in plasma chylomicrons, VLDL, and HDL. ApoA5 is thought to facilitate the interaction of endothelial heparan sulfate with apoCII on triglyceride-rich lipoproteins and the interaction of apoCII with LPL on the vascular endothelium. Several families with apoA5 deficiency have severe hypertriglyceridemia. These defects may be more prevalent among Asians (Pullinger et al 2008).
The apolipoprotein A5 (ApoA5) gene plays an important role in determining plasma triglyceride concentrations in humans (Kao et.al. 2003). This gene is located proximal to the apolipoprotein gene cluster on chromosome 11q23. It determines plasma triglyceride (TG) levels by both being a potent stimulator of lipoprotein lipase mediated TG hydrolysis and a inhibitor of the hepatic VLDL-TG production rate, without affecting the VLDL-apoB production rate. Defects in APOA5 are a cause of susceptibility to familial hypertriglyceridemia, a common inherited disorder in which very low density lipoprotein (VLDL) levels are elevated in plasma. Defects in APOA5 are also a cause of hyperlipoproteinemia type 5, characterized by increased amounts of chylomicrons and very low density lipoprotein (VLDL) in the plasma. Numerous conditions cause this phenotype, including insulin-dependent diabetes mellitus, contraceptive steroids and alcohol abuse.
In our patient gestational diabetes with insulin resistance contributed further to the genetic susceptibility to raise triglyceride levels. Mild hypertriglyceridaemia, with up to four fold increases in plasma triglycerides due to elevated oestrogen and human placental lactogen levels, is common in the second and third trimesters of pregnancy (Lain et.al.2007, Knopp et.al.1986, Herrera 2002). Oestrogens increase triglyceride-rich lipoprotein secretion and suppress hepatic lipase activity. Increased human placental lactogen leads to relative peripheral insulin resistance with suppressed plasma lipoprotein lipase activity, enhanced plasma CETP activity and enhanced free fatty acid flux to the liver.
Both chylomicron and VLDL remnant particles acquire apoE before they can bind to hepatic receptors for uptake and degradation or further processing to LDL. The ApoE gene has three common alleles of which the ApoE2 allele product has reduced receptor affinity (Mahley, 2000). Hence this patient’s ApoE2 heterozygosity also contributed to hypertriglyceridaemia.
Dietary measures are the first step in management of LPL deficiency and consist of reducing the dietary intake of fat to approximately 25% of total daily caloric intake. This fat should be provided in the form of medium-chain triglycerides which are transported to the liver without prior chylomicron packaging, thus excluding the need for LPL. One case report describes delivery of a healthy infant to a mother whose chylomicronaemia secondary to LPL deficiency was managed with extreme dietary fat restriction to less than 2 g per day during the second and third trimesters (Tsai et.al. 2004). Our patient responded modestly to fat restriction and hence additional treatment was necessary.
Omega-3 fatty acid capsules e.g. Omacor® and Maxepa® contain eicosapentaenoic acid ethyl ester and docosahexaenoic acid ethyl ester which are essential fatty acids. Doses of 3 to 4 grams daily suppress hepatic lipogenesis and enhance fatty acid oxidation (Davidson, 2006) and may directly stimulate LPL to enhance removal of triglyceride-rich lipoproteins (Khan et.al. 2002). Thus they reduce serum triglycerides by up to 30% in healthy persons (Skulas-Ray et.al. 2011). Treatment with omega-3 fatty acids in LPL-deficiency has rarely been reported. There was a beneficial therapeutic response to medium-chain triglycerides and omega-3 fatty acids in a patient with the familial chylomicronaemia syndrome (Rouis et.al. 2011). Three case reports of gestational hypertriglyceridaemia treated with omega-3 fatty acids leading to a good outcome are reviewed by Goldberg and Hegele (2012). Our results indicate a good response, free of side effects, to this product during pregnancy and from 17 years after pregnancy onwards in combination with other lipid regulating drugs. From these reports an adverse pregnancy outcome from the use of omega-3 products appears unlikely.
The introduction of colestyramine to the treatment during pregnancy led to a severe increase in serum triglycerides and subsequent pancreatitis. Colestyramine interrupts the enterohepatic circulation of bile and is associated with increased production of VLDL, possibly due to enhanced hepatic triglyceride synthesis (Angelin et.al. 1978). Hepatic production of cholesterol and bile acid formation are also stimulated, leading to increased synthesis of VLDL from the newly generated cholesterol.
Following the pregnancy, fibrates and statins were initially effective in managing the hyperlipidaemia. At twenty years after pregnancy a combination of high doses of statin, fibrate and omega-3 fatty acids was required. This additional requirement was probably due to the onset of type 2 diabetes mellitus aggravating the dyslipidaemia.
Recently, gene replacement therapy has been approved in Europe for use in familial LPL deficiency with severe or multiple pancreatitis attacks despite dietary fat restrictions. Functional copies of the gain of function LPL gene variant are introduced into the muscle tissue of patients using an adenoviral-associated viral vector packaged in a capsid that directs the vector to skeletal muscle (Gaudet et.al. 2012). In two clinical trials, LPL deficient patients were administered intramuscular injections followed by several weeks of immunosuppressive drugs to reduce the immune responses to the viral capsid. The treatment proved successful in reducing both the serum triglyceride levels for up to 12 weeks after injection as well as the incidence of pancreatitis attacks for up to 2 years after treatment. No significant side-effects have been described. An assessment of the use of gene therapy for LPL and apoA5 deficiency in pregnancy is awaited (Goldberg and Hegele, 2012 ). Our patient may be considered for gene therapy in the future if her hyperlipidaemia becomes refractory to standard medication with progression of diabetes.
Conclusion
The coexistence of a heterozygous LPL gene mutation, homozygous and heterozygous ApoA5 gene mutations, gestational diabetes and ApoE2 heterozygosity resulted in marked hypertriglyceridemia and acute pancreatitis during a pregnancy. Treatment of hypertriglyceridaemia with omega-3 fatty acids was effective during this pregnancy, before the introduction of colestyramine. After pregnancy she initially responded to monotherapy with fibrates but later required a combination of lipid regulating drugs. Colestyramine should not be used in pregnancy as it may raise triglyceride levels and cause acute pancreatitis. Gene replacement therapy for LPL or apoA5 deficiency provides hope as a future effective therapy for this rare serious inherited condition.
Figure 1. Serum Lipids during Pregnancy from 13 Weeks Gestation Showing Responses to Very Low Fat Diet, Omega-3 Fatty Acids and Colestyramine Therapy up to Time of Caesarean Section at 29 Weeks Gestation
Table 1: Serum Lipids and Medication Used over 21 Years after Pregnancy. Key: TC = Total Cholesterol, TG = Triglycerides, HDL-c = High Density Lipoprotein Cholesterol, F = Fenofibrate, A = Atorvastatin, Om = Omega-3 Fatty Acids
Acknowledgements:
Thanks to
– Drs. Sheila Mallya and David Wile for contributions to patient investigations, management and comments on the manuscript.
– Scientific staff at the Regional Genetics Laboratories, Belfast City Hospital Trust, Belfast for genetic analysis and comments on the results.
– The patient who has given fully informed written consent for publication of this report.