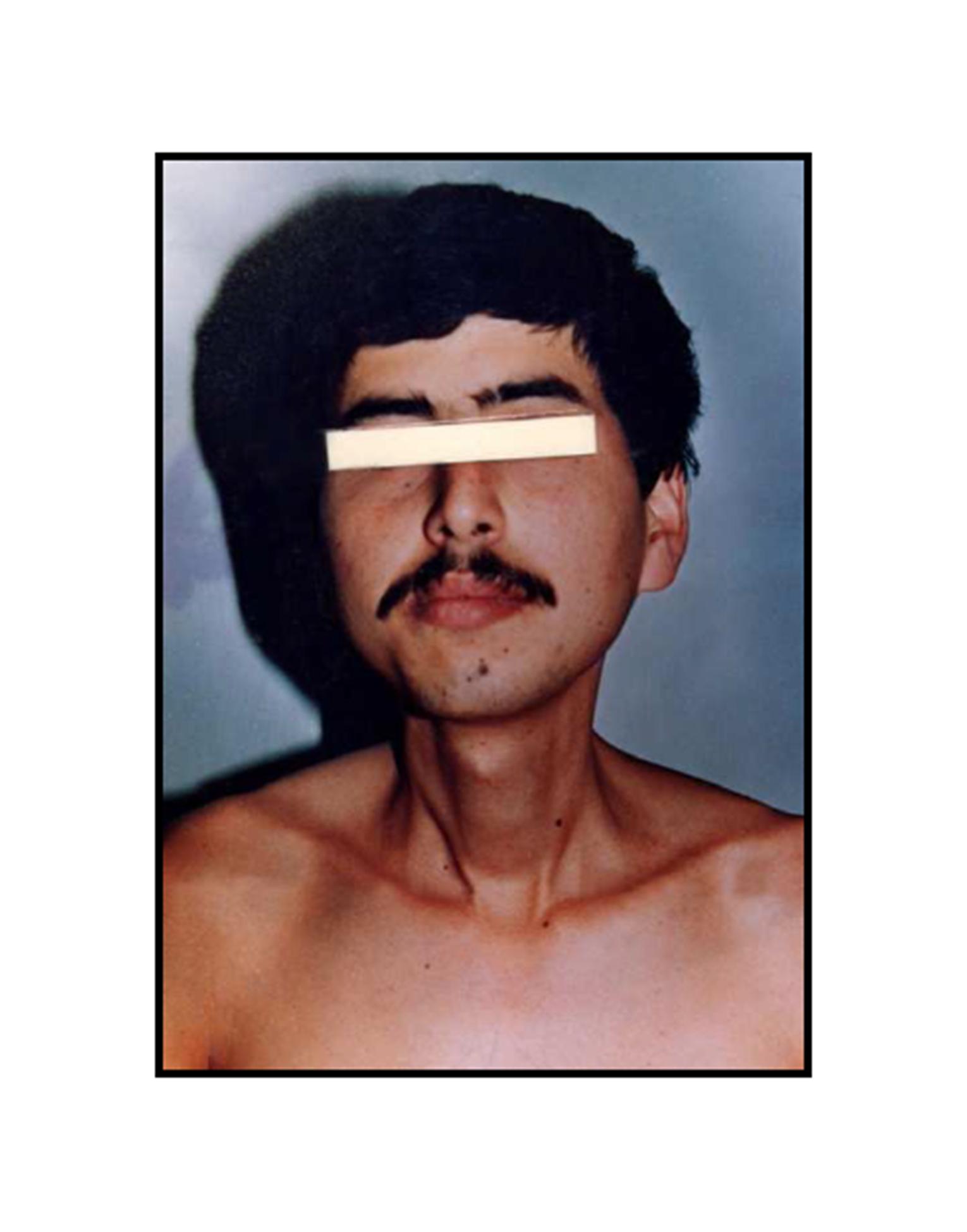
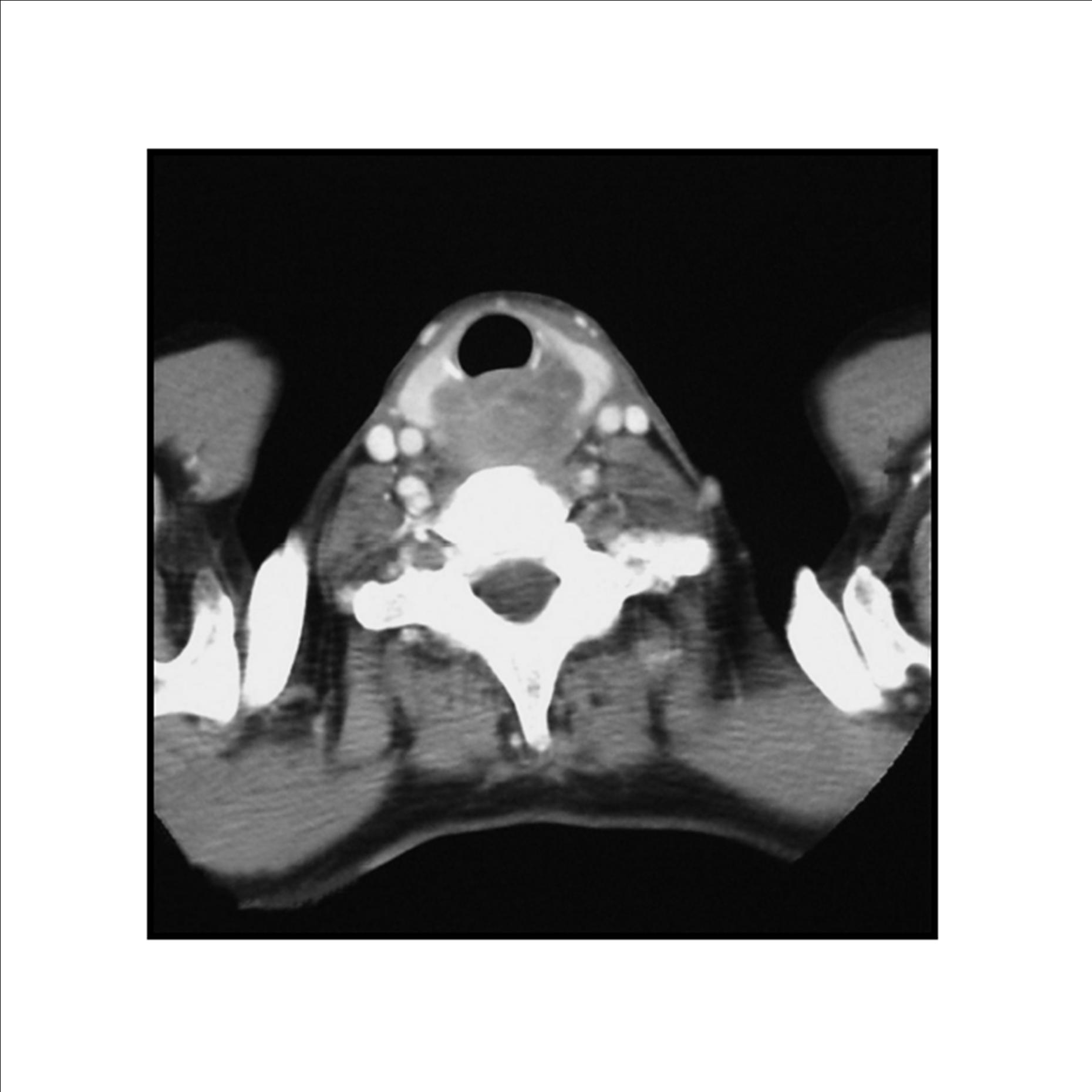
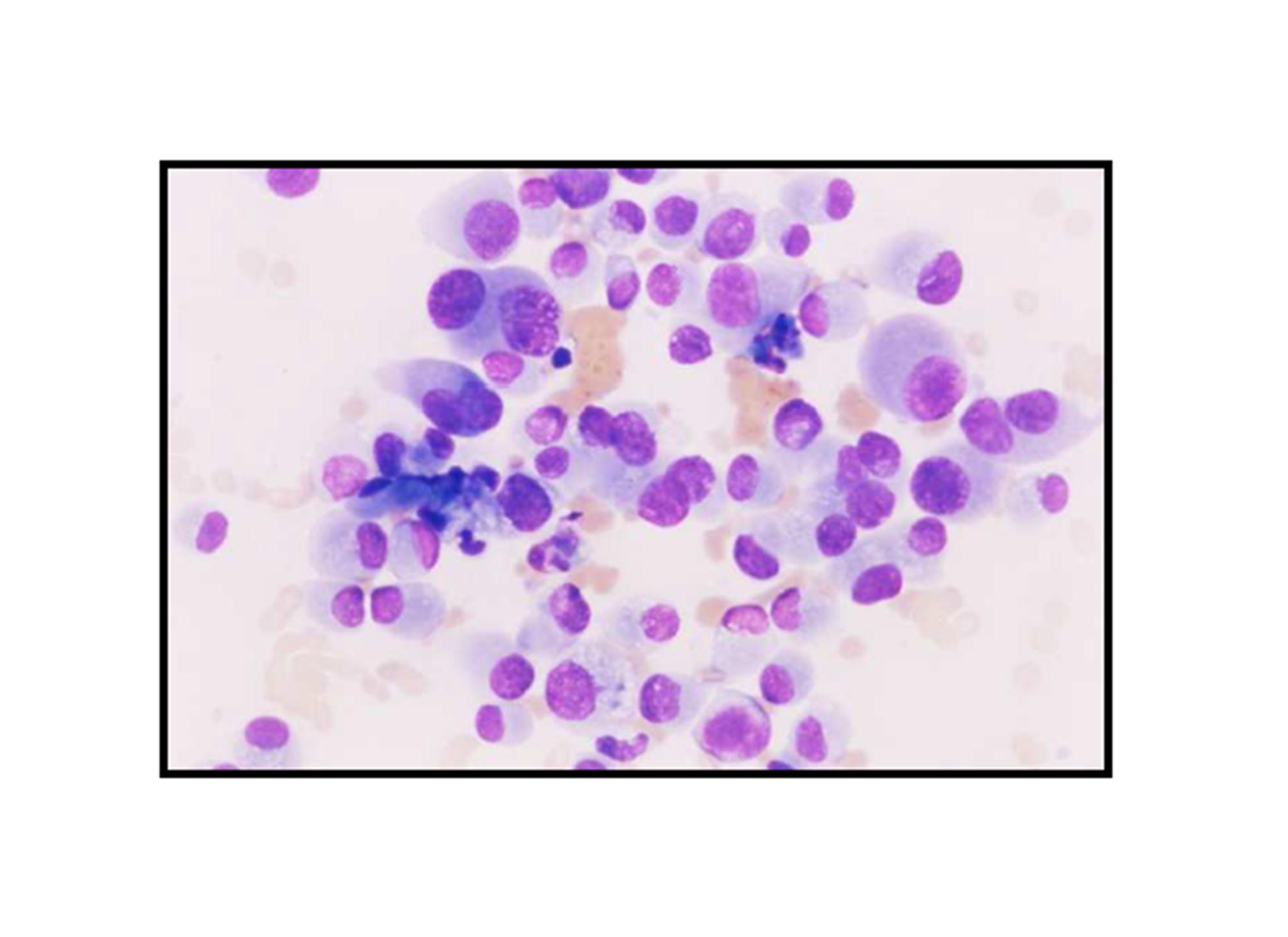
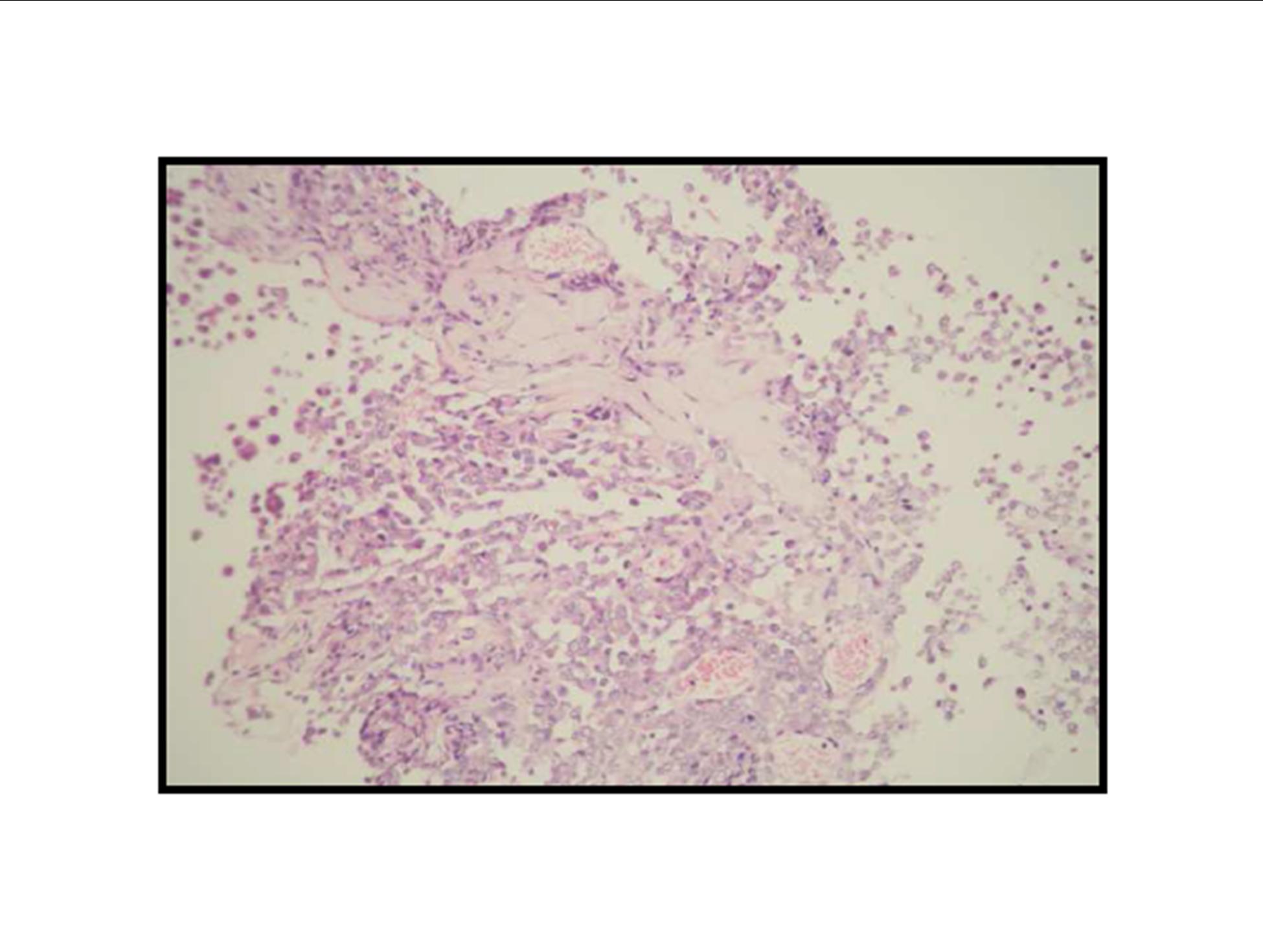
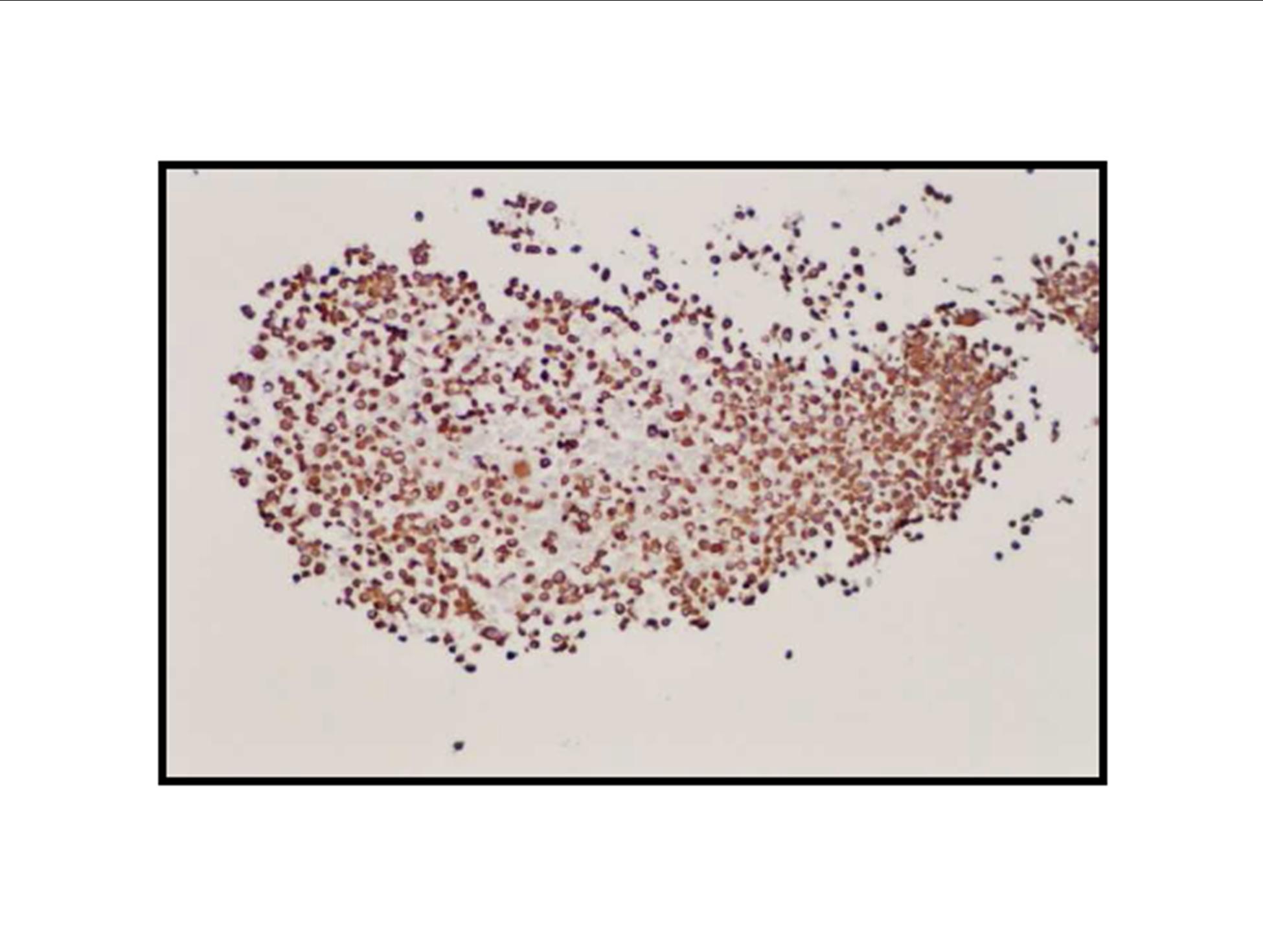
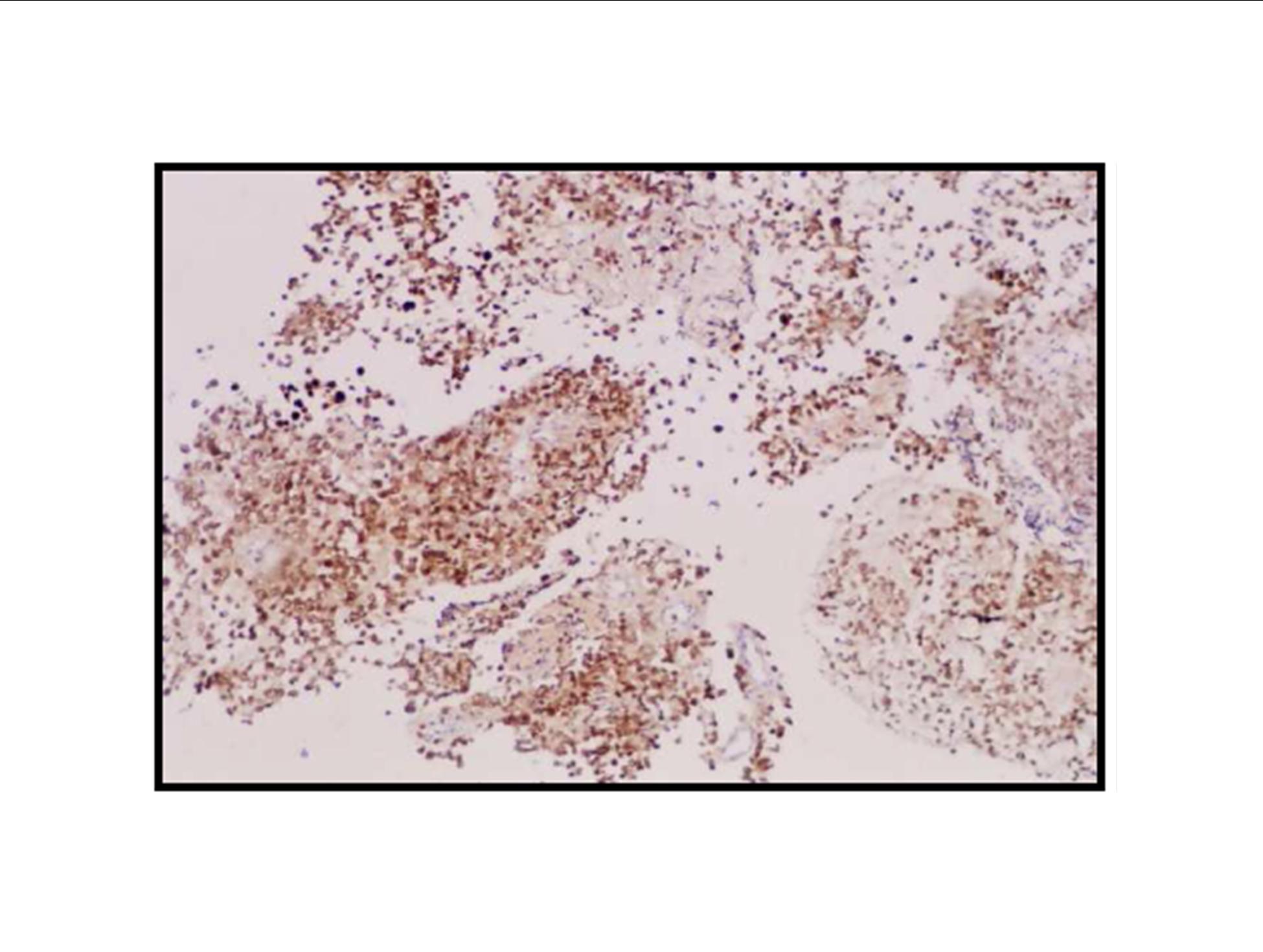
Pathologic diagnosis was epithelioid malignant schwannoma. The patient received a further four cycle ifosfamide + adriamycin combination chemotherapy, but remission could not be obtained. The patient died with progression of his disease 6 months after diagnosis.
Discussion
MPNSTs form a heterogenous group of neoplasms, originating from the different cells of the nerve sheath including Schwann cells, perineural cells, and fibroblasts (Layfield, 2002). The diagnosis of MPNSTs requires at least one of the following criteria: (1) a tumor arising from a peripheral nerve usually a large nerve trunk, (2) a tumor originating from a pre-existing benign nerve sheath tumor, (3) occurrence in a patient with neurofibromatosis or (4) definitive ultrastructural evidence of Schwann cell differentiation. In the absence of these features, it is possible to reach the correct diagnosis with the presence of classic morphologic features and characteristic immunohistochemistry findings (Weiss and Goldblum, 2001; Dodd et al., 1997). MPNST is a malignant mesenchymal neoplasm usually involving the extremities and the trunk. These tumors rarely involve the upper rodigestive system (Weiss and Goldblum, 2001; Layfield, 2002).
There is a limited number of studies on the cytological features of EMPNSTs.. The Cytologic features of this tumor were first described by Molenaar et al. in 1989. Cytologically, these tumors usually consist of loosely cohesive clusters or dispersed large polygonal to plasmacytoid or epithelioid cells with prominent melanoma-like nucleoli. The tumor cells have generally moderate to large and finely granular cytoplasm with occasional microvacuolisation. Mitotic figures, prominent nuclear pleomorphism, and binucleation are other characteristic features (Dodd et al., 1997; Reis et al., 2002). Cyto/histomorphologic findings alone are not sufficient for EMPNST diagnosis. Obtaining a correct diagnosis is easier in patients with NF1 or when such a tumor arises from a major nerve or preexisting neurofibroma. In the absence of a suggestive clinical setting, on the basis of cyto/histomormorphologic features alone, the diagnosis of EMPNST is vague and immunocyto/histochemical and ultrastructural studies are essential to make a correct diagnosis and to exclude other possibilities. Strong immunostaining with S-100 protein, vimentin, GFAP or CD57 (Leu-7) are characteristic features for EMPNST (Dodd et al., 1997; Reis et al., 2002; Kwon et al., 2002).
A swan-like neck was seen in the patient 12 years later after the treatment and epithelioid malignant schwannoma developed in the irradiated area 30 years later after the radiotherapy. The patient could not achieve remission and died although four cycle ifosfamide + adriamycin combination chemotherapy were given. Adamson et al. (2004) reported two patients with MPNST of the spine after radiotherapy for Hodgkin’s lymphoma and despite prompt surgical resection MPNSTs exhibited an aggressive behavior. The prognosis in the patients with MPNSTs is still poor.
On the other hand, in addition to chemoradiotherapy, genetic predisposing factors such as Li-Fraumeni syndrome, neurofibromatosis, genetic retinoblastoma have a potential for developing secondary neoplasms. It is also important to note that, genetic susceptibility may also play a role (Boice, 1996). Genomic analysis revealed alterations in cell cycle, repair, while detoxification and stress response pathways are suggested to be involved in the development of HL and in the occurrence of second neoplasias in these patients (Lorenzo et al., 2009).
M’Kacher et al. (2007) investigated the relationship between telomere shortening in peripheral blood lymphocytes, increased chromosome abnormalities, radiation sensitivity and secondary cancers. Prior to treatment, patients with HL showed age-independent shorter telomeres, increased spontaneous chromosomal abnormalities and increased in-vitro radiation sensitivity. After treatment, telomere shortening was associated with cytogenetic profiles characterized by the persistence of complex chromosomal rearrangement and clonal aberrations. Patients with second cancers were characterized by markedly short telomeres, the presence of complex chromosome rearrangements and increased in vitro radiation sensitivity. An intimate relationship between pre-treatment telomere shortening, chromosomal instability, radiation sensitivity and occurrence of second cancers was determined in the cases (M’Kacher et al., 2007).
The age at treatment has a major effect on risk of second malignancy after therapy for HL. A cohort of 5,519 patients with HL treated during 1963-1993 was evaluated and followed-up for second malignancy. Three hundred twenty-two second malignancy occurred. Relative risks of cancers and of leukemia increased significantly with younger age at first treatment. However, absolute excess risks and cumulative risks of cancers and leukemia were greater at older ages. Although absolute excess risks are greater for older patients, relative risks of several important malignancies are much greater for patients who were treated when young (Swerdlow et al., 2000).
Conclusion
The clinicians should be aware of and alert for SMNs, especially in the patients treated with radiotherapy. Monitoring for the detection of SMNs in the survivors of childhood cancer necessitates a good collaboration between pediatric and adult oncology departments.
References
1. Adamson, D. C., Cummings, T. J. and Friedman, A.H. (2004) “Malignant peripheral nerve sheath tumor of the spine after radiation therapy for Hodgkin’s lymphoma,” Clinical Neuropathology, 23 (5) 245-255.
2. Bhatia, S., Robison, L. L., Oberlin, O., et al. (1996) “Breast cancer and other second neoplasms after Childhood Hodgkin’s disease,” The New England Journal of Medicine, 334 (12) 745-751.
Publisher – Google Scholar
3. Boice Jr., J.D. (1996) Cancer following irradition in childhood and adolescence, In: Green, D. M. and D’Angio, G. J., editors. “Mutagenic and carcinogenic effects of treatment for childhood cancer,” Medical and Pediatric Oncology, (suppl 1) 29-35.
4. Dodd, L. G., Scully, S. and Layfield, L. J. (1997) “Fine-needle aspiration of epithelioid malignant peripheral nerve sheath tumor (epithelioid malignant schwannoma),” Diagnostic Cytopathology, 17 (3) 200-204.
Publisher – Google Scholar
5. Kwon, M. S., Lee, S. S. and Ahn, G. H. (2002) “Schwannomas of the gastrointestinal tract: clinicopathological features of 12 cases including a case of esophageal tumor compared with those of gastrointestinal stromal tumors and leiomyomas of the gastrointestinal tract,” Pathology Research and Practice, 198 (9) 605-613.
Publisher – Google Scholar
6. Layfield, L. J. (2002) Cytopathology of Bone and Soft Tissue Tumors, Oxford University Press, New York, USA.
7. Lorenzo, Y., Provencio, M., Lombardia, L., et al. (2009) “Differential genetic and functional markers of second neoplasias in Hodgkin’s disease patients,” Clinical Cancer Research, 15 (15) 4823-4828.
Publisher – Google Scholar
8. M’kacher, R., Bennaceur-Griscelli, A., Girinsky, T., et al. (2007) “Telomere shortening and associated chromosomal instability in peripheral lymphocytes of patients with Hodgkin’s lymphoma prior to any treatment are predictive of second cancers,” International Journal of Radiation Oncology Biology Physics, 68 (2) 465-471.
Publisher – Google Scholar
9. Meadows, A. T., Baum, E., Fossati-Bellani, F., et al. (1985) “Second malignant neoplasms in children:An update from the late effects study group,” Journal of Clinical Oncology, 3 (4) 532-538.
Google Scholar
10. Molenaar, W. M., Ladde, B. E., Scraffordt Kroops, H., et al. (1989) “Two epithelioid malignant schwannomas in a patient with neurofibromatosis. Cytology, histology and DNA-flow-cytometry,” Pathology Research and Practice, 184 (5) 529-534.
Publisher – Google Scholar
11. Reis Filho, J. S., Pope, L. Z., Balderrama, C. M., et al. (2002) “Epithelioid malignant peripheral nerve sheath tumour: case report and review of the previously published cases,” Cytopathology, 13 (1) 54-63.
Publisher – Google Scholar
12. Swerdlow, A. J., Barber, J. A., Hudson, G. V., et al. (2000) “Risk of second malignancy after Hodgkin’s disease in a collaborative British cohort: the relation to age at treatment,” Journal of Clinical Oncology, 18 (3) 498-509.
Google Scholar
13. Weiss, S. W. and Goldblum, J. R. (2001), Malignant tumors of the peripheral nerves, In: Weiss, S. W. and Goldblum, J. R., editors. Soft Tissue Tumors. 4th ed. St Louis, CV Mosby, 1209-1263.