From the Department of Endocrinology, (J-O. J., J. A), Department of Clinical Physiology (K.C, A.P.), Department of Clinical Chemistry, Institute of Biomedicine (J-E. M.), Sahlgrenska University Hospital and Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Received date: 19 May 2014; Accepted date: 6 August 2014; Published date: 22 January 2015
Academic Editor: Erik Ilsø Christensen
Cite this Article as:
Jan-Ove Johansson, Jarl Ahlmén ,Anita Persson, Kenneth Caidahl and Jan-Eric Månsson (2015), “Adult Fabry patients in Sweden - Baseline data and Outcome of Four-Year Enzyme Replacement Treatment in a Male Cohort without Proteinuria ", JMED Research, Vol. 2015 (2015), Article ID 373829, DOI: 10.5171/2015.373829
Background: Fabry disease is a rare X-linked lysosomal storage disease caused by defective activity of the lysosomal enzyme α-galactosidase A. The objective was to study the baseline characteristics of Fabry patients in Sweden, Furthermore, to study the effect of 4 years of ERT (enzyme replacement therapy) in an open study of a subcohort of Fabry men with no or mild proteinuria (<1 gram/day). Patients and methods: Thirty-four Swedish Fabry patients were enrolled in the baseline study during a 10 year period. We present baseline data for both men and women. The effect of 4 years of ERT was studied in an open study of a subcohort of 12 Fabry men with no or mild proteinuria. In the first 12-18 months this subcohort received the licensed dose of agalsidase alfa (Replagal®) 0.2 mg/kg body weight every other week and the men were then switched to a reduced dose of agalsidase beta (Fabrazyme®), 0.5 mg/kg body weight every other week. Results: Swedish Fabry patients showed a similar prevalence of classical symptoms compared to Fabry patients from other countries, except for a lower prevalence of renal complications in the Swedish women. The 4 years of ERT of men with no or mild proteinuria resulted in maintained kidney function. In addition, there was no increase in left ventricular mass. Conclusion: The study indicates that ERT stabilizes kidney function in Fabry patients with no or mild proteinuria and has a cardioprotective effect.
Fabry disease is a rare X-linked lysosomal storage disease caused by a mutation of the gene that encodes the enzyme α-galactosidase A. This enzyme deficiency is life-threatening and causes a progressive accumulation of glycosphingolipids, mostly globotriaosylceramide (Gb3), within many cell types including the vascular endothelium (Desnick et al., 2001). Mostly men are affected, although carrier (heterozygous) women can have serious disease manifestions. Women can range from being asymptomatic to having severe symptoms which is likely related to non-random X-chromosomal inactivation during embryonic development of the female (Lyon 1961, Mac Dermot et al., 2001). The incidence of Fabry disease in men has been estimated at 1:117,000 births (Meikle et al., 1999), although screening studies of newborn children have suggested a much higher incidense (Spada et al., 2006).
Clinical manifestations usually become apparent around the age of 8-10 years in affected boys and about 2-5 years later in girls. The early symptoms include burning neuropathic pain, mostly in the hands and feet during work or fever (acroparesthesia), hypohidrosis, skin lesions (angiokeratoma corporis diffusum) and corneal opacities (cornea verticillata) (Desnick et al., 2001), albuminuria and proteinuria.
In Fabry disease, progressive accumulation of glycosphingolipids starts early in life and continues for decades. Over time, the accumulation of glycosphingolipids also leads to progressive kidney failure, hypertrophic cardio and cerebrovascular disease. By the third to fifth decade more serious complications from these organs typically occur, ultimately leading to high morbidity and premature mortality (Desnick et al., 2001, Mac Dermot et al., 2001). In addition, patients may suffer from hearing loss and lymphedema. We started the ERT of the Fabry patients very early in the treatment era in Europe and there was a great uncertainty about the dosing of the two enzymes. 2001 EMEA (European Medicines Agency) had approved the dose 0.2 mg/kg for agalsidase alfa (Replagal®) and 1.0 mg/kg body weight every other week for agalsidase beta (Fabrazyme®) (Desnick 2004, Schaefer et al., 2009). Soon after the approval a study showed that the two products had virtually identical biochemical profiles (enzyme activity per mg protein, uptake in fibroblasts, biodistribution and antibody cross-reactivity) (Lee et al., 2003). In order to increase the dose of enzyme and to individualise the dosing, we switched our patients from agalsidase alfa 0.2 mg/kg body weight every second week to a dose of agalsidase beta of 0.5 mg/kg body weight every second week, thereby increasing the given dose of enzyme 2.5 times. After our study and after publication of the study of Banikazemi et al., the dose of agalsidase beta was increased to 1.0 mg/kg (Banikazemi et al., 2007).
With our study we wish to give a survey of the past and present situation concerning Fabry disease in Sweden. The aim of the study was to report the baseline characteristics of Fabry patients. Furthermore, the effect of 4 years of ERT was evaluated in an open study of a subcohort of Fabry men with no or mild proteinuria (<1 gram/day).
Subjects and methods
Patients were diagnosed with Fabry disease by demonstration of a reduction of α-galactosidase A activity in leukocytes and by DNA mutation analysis. Thirty-four Caucasian Swedish Fabry patients, 19 men and 15 women from 15 different families, were studied at baseline during the years 2000-2009. In addition, 12 Fabry men with no or mild proteinuria were studied during 4 years of ERT in a prospective study.
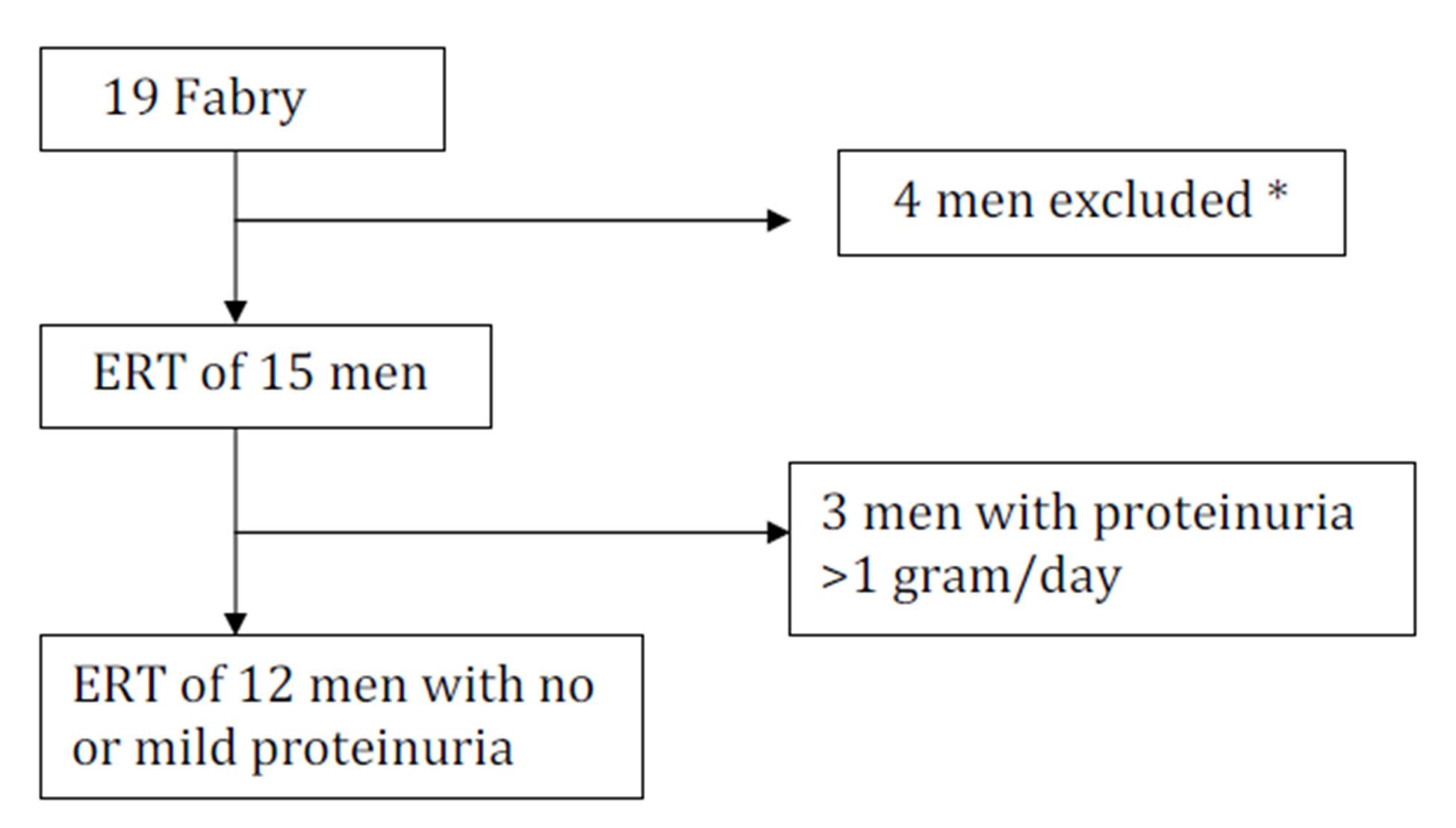
Only 15 of the original 19 men could be followed during 4 years of ERT (Fig. 1). All the 19 men started ERT in the beginning of the study.
The Fabry patients’ complete medical histories were obtained and they underwent at least yearly physical examinations.
Biochemical Assays Venous blood samples were taken for the determination of the mutation in the α-galactosidase gene, α-galactosidase activity and Gb3 levels in plasma. Blood samples were drawn in 6-mL Vacutainer tubes. BD, Belliver Industrial Estate, Plymouth, UK.
The activity of a-galactosidase A in leukocytes and fibroblasts was measured using a fluorimetric assay with 4-methylumbelliferyl-a-D-galactopyranoside as substrate (Svennerholm et al., 1979) in the presence of 0.1 M N-acetylgalactosamine as inhibitor of a-N-acetylgalactosaminidase (Mayes et al., 1981). The concentration of Gb3 in plasma and urinary sediment was quantified by densitometric evaluation after HPTLC and orcinol detection (Kyllerman et al., 1990).
Renal function by glomerular filtration rate (GFR) measurement
GFR measured by iohexol clearance was performed yearly as a clinical routine method, as previously described (Nilsson-Ehle et al., 1994). The normal ranges are shown in Table 4.
Doppler-echocardiography
A complete echocardiographic investigation was performed yearly as previously described (Waagstein et al., 1989, Persson et al., 2010). Left ventricular dimension was measured by M-mode, and the LV ejection fraction (LVEF) was estimated using the Teichholtz formula. LV mass was calculated from the parasternal M-mode projection using the ASE formula as LVmass = 0.8 (1.04 ([LVd + PWd + IVSd]3) -[LVd]3) + 0.6 g, where LVd, PWd and interventricular septum diastole (IVSd) are the diastolic diameters of the left ventricle, posterior wall and interventricular septum.
Left ventricular (LV) hypertrophy was defined as LV mass >261 g in men and >193 g in women. We regarded the normal range for LV diastolic dimension (LVd) to be 4.6-6.0 cm in men and 4.1-5.1 cm in women. For posterior wall (PWd) ≤1.1 cm in both genders and for interventricular septum (IVSd) ≤1.2 cm in men and ≤1.1 cm in women were regarded normal. For LVEF the reference level used was ≥56% in men and ≥54% in women.
Magnetic resonance imaging (MRI)
MRI of the brain was performed using an Intera/Achieva 1.5 Tesla (Philips, Best, and The Netherlands) with intravenous gadolinium-infusion, at baseline and after 2 and 4 years, respectively.
Eudiometry
A conventional audiometry via the Department of Audiology was used for the evaluation of hearing.
Ethical consideration
Informed written consent was provided by all subjects and the study was approved by the Ethics Committee of the Medical Faculty at Gothenburg University.
Statistics
Differences between baseline and after 2 and 4 years of treatment, respectively were tested using a non-parametric test: The Wilcoxon test for paired differences (Wilcoxon signed rank test). Mann Whitney’s U-test was used to tests differences between subgroups in the Fabry cohort. Values are given as means and standard errors of the means (SEM).
The ERT study
The design of the study was to study the effect of 4 years ERT on GFR in an open study of a subcohort of Fabry men with no or mild proteinuria (<1 gram/day),
During the first 12-18 months of the study agalsidase alfa (Replagal®) 0.2 mg/kg body weight every other week was used and the patients were then switched to half the licensed dose agalsidase beta (Fabrazyme®), i.e. 0.5 mg/kg body weight every other week up to 48 months.
After the 4-year study, the dose of agalsidase beta was increased to the licensed dose of 1.0 mg/kg body weight every other week (Banikazemi et al., 2007). Results
Thirty-four Fabry patients, 19 men and 15 women, from 15 different families, accepted to participate in the study.
The patients were enrolled in the study during the 10 year period 2000-2009. In 2 of the Fabry families, one woman aged 53 and one man aged 55 died as a consequence of their severe Fabry disease during this period, and hence, baseline data are not available.
Besides the 15 Fabry families in our study we are aware of 3 additional families that are receiving care at other hospitals in Sweden. Two of these families, with only one patient in each, have recently immigrated to Sweden from two other European countries. None of these families has been included in the study. In addition to the 18 Fabry families mentioned above, another 3 Fabry families have existed in Sweden during the last decades. Two male Fabry patients died 1991, aged 48 and 32 respectively. One woman belonging to a third Fabry family died aged 51 of cerebrovascular disease in 2009. Today, no known Fabry patients remain in these 3 families in Sweden.
As a summary, we are aware of 18 different Fabry families in Sweden for the moment. In these families there are 51 patients with Fabry disease: 18 adult males, 1 boy aged 13, 1 boy aged 3 and 31 women or girls. Twenty four of them are receiving ERT: seventeen men, the 13 year-old boy and 6 women
Baseline data
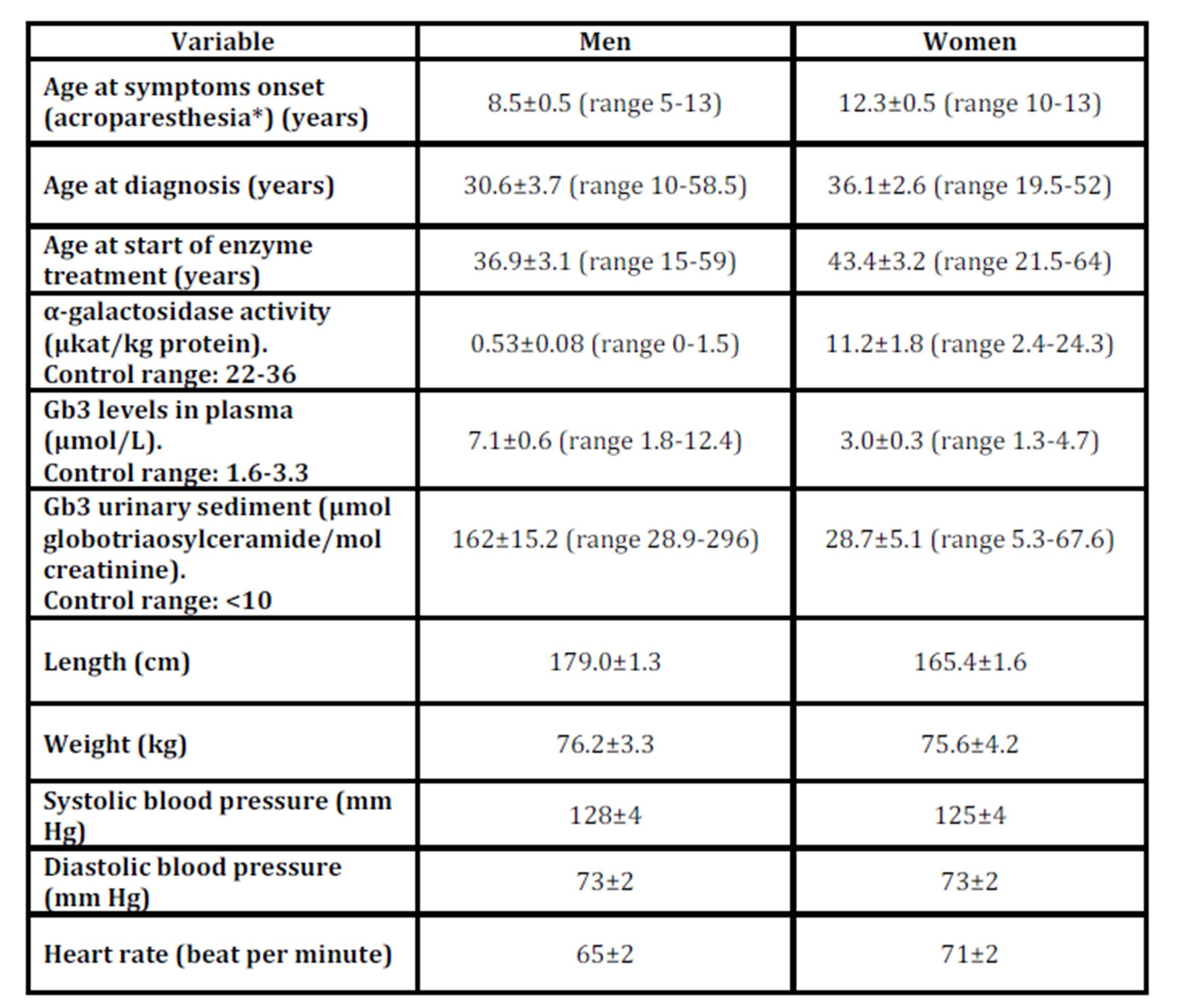
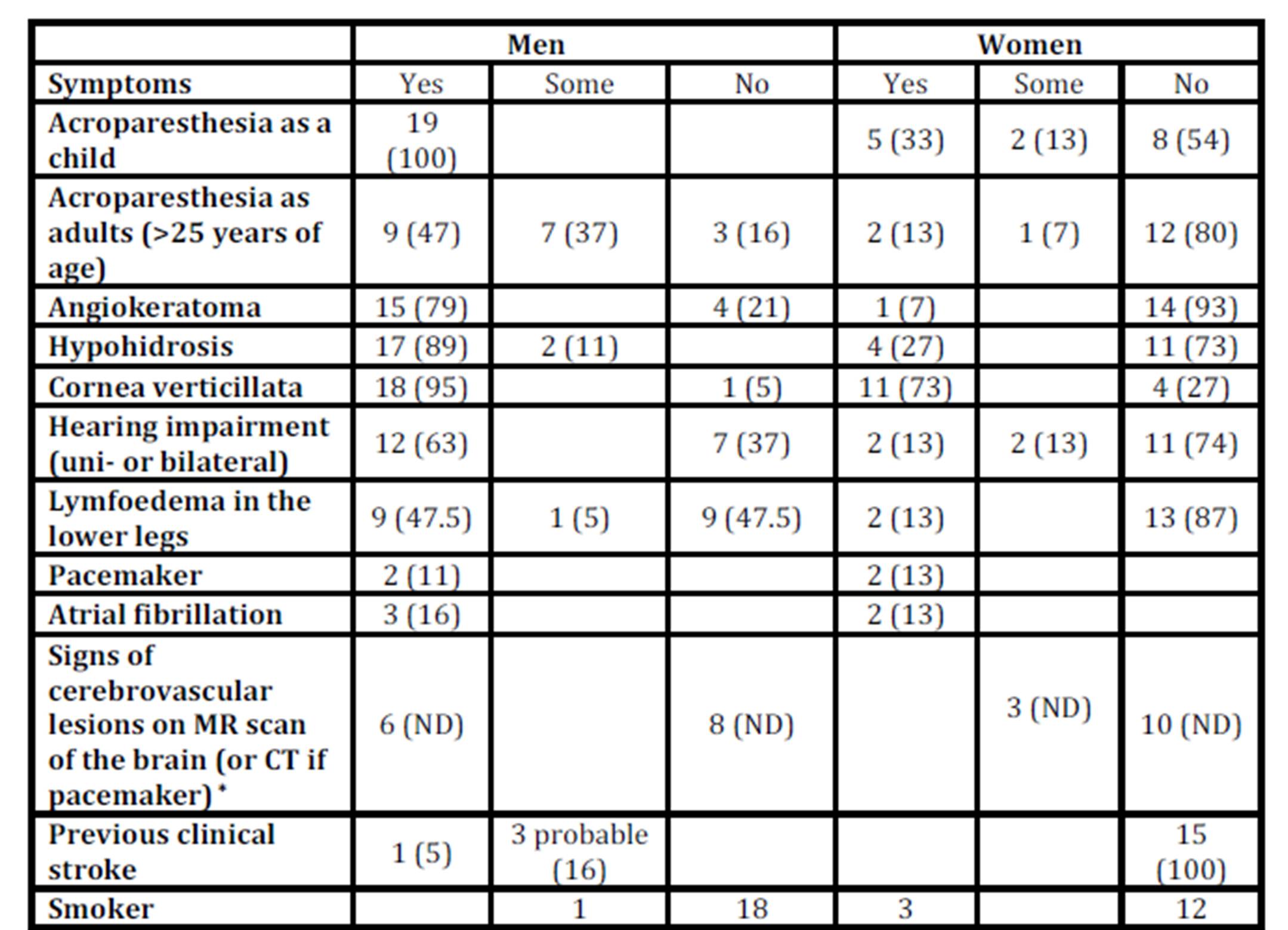
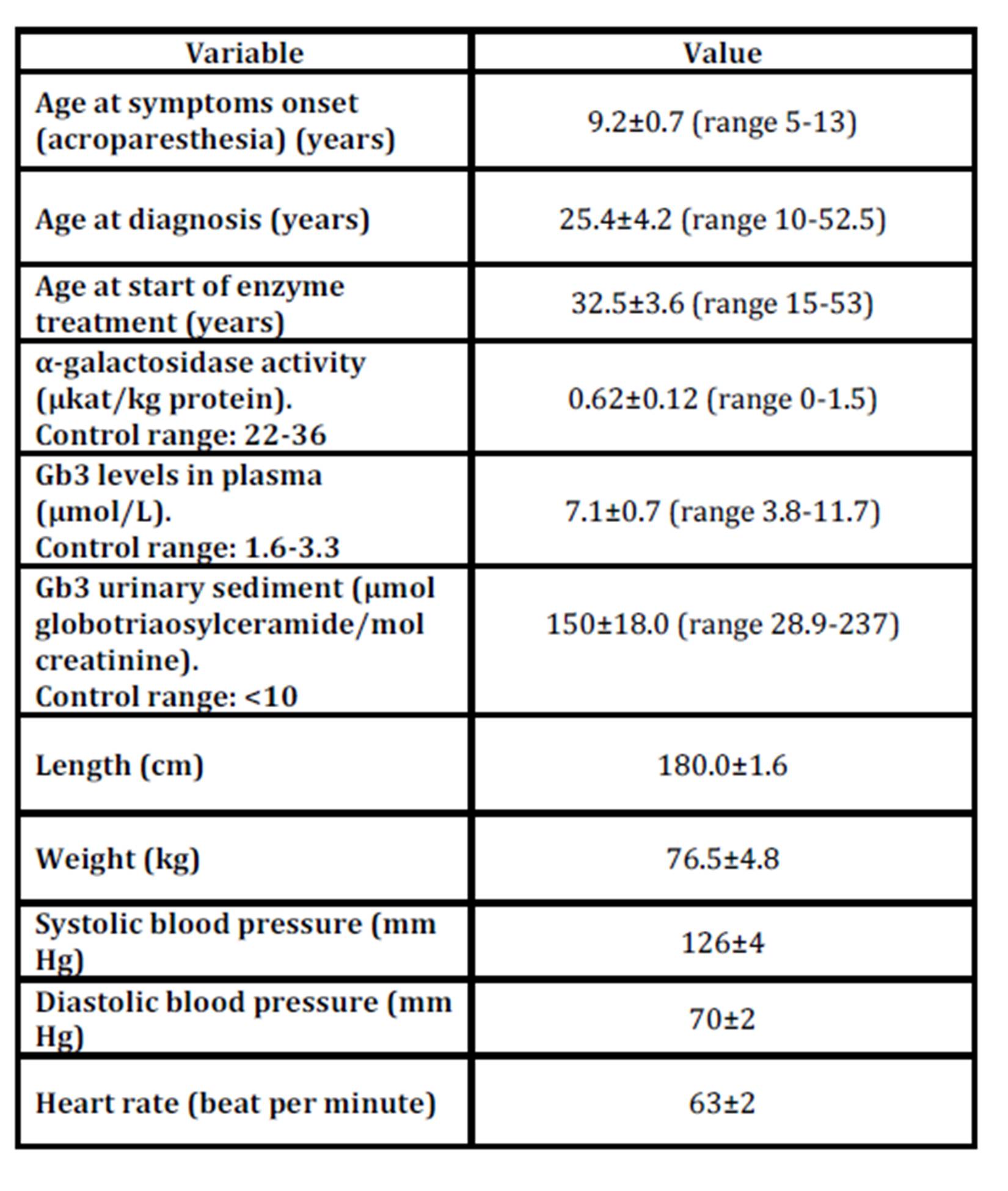
The baseline characteristics are shown in Table 1 and the symptoms in Table 2 of the 19 men and the 15 women with Fabry disease.
Table 1: Baseline characterisics in 19 men and 15 women with Fabry disease
* Men n=19, women n=7
Table 2: Symptoms of Fabry disease in 19 men and 15 women
Number of patients, Percentages of the own gender in parentheses
* Five Fabry men and 2 women could not be investigated with an MR scan of the brain at baseline (percentages not determined, ND, in the table).
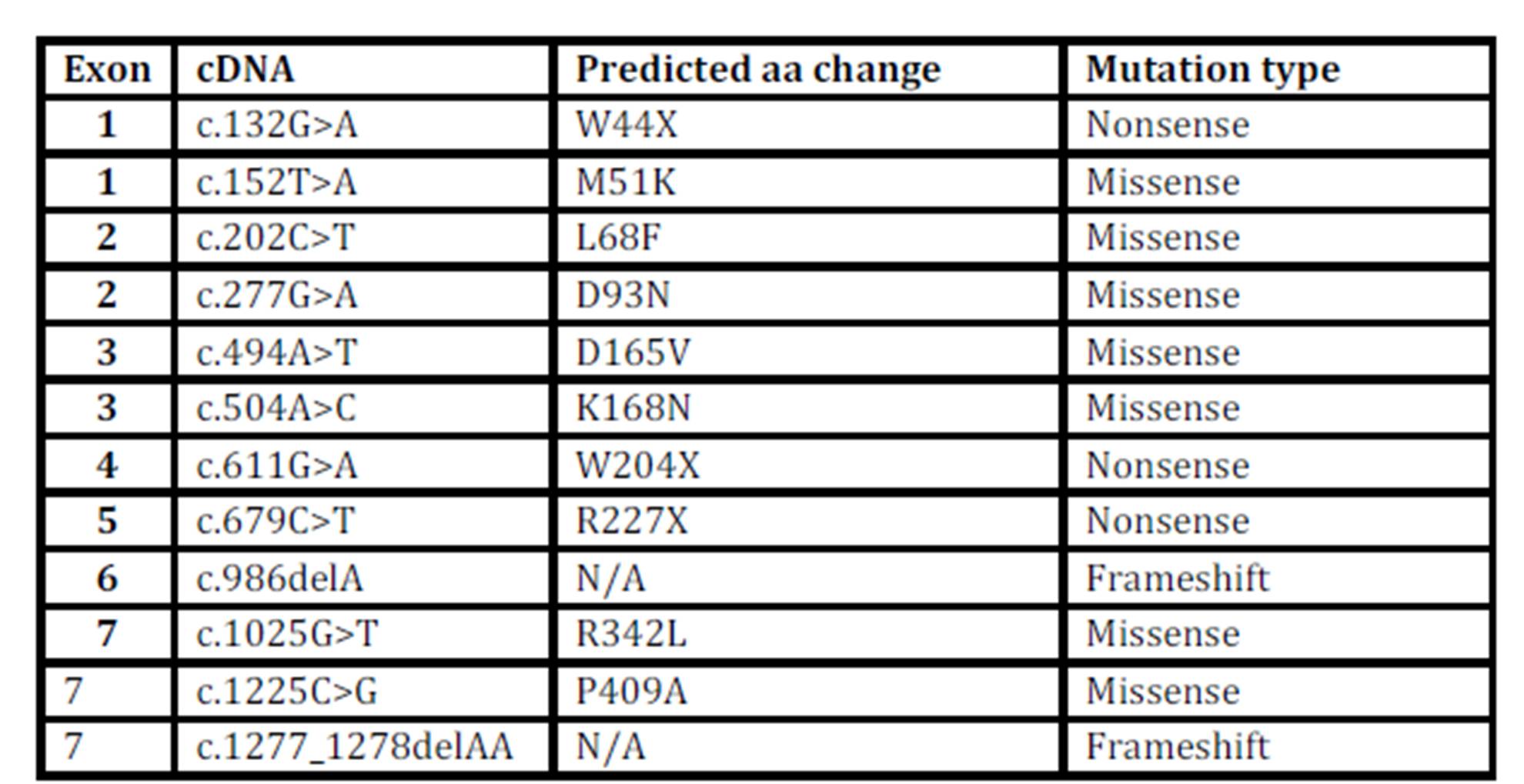
The different mutations of the α-galactosidase gene that were found in the Swedish Fabry families are presented in Table 3.
Table 3: The mutations
α-galactosidase A sequence variants identified in the Swedish Fabry families.
Renal function in Fabry men
Renal function and proteinuria in the 19 Fabry men at baseline
Seven of the 19 male patients (37%) had reduced GFR, 54-77 ml/min per 1.73 m2, at baseline (age 43.4±3.5, range 27-53 years) while 12 patients (63%) had normal GFR (age 33.0±4.2, range 15-59 years). The difference in mean age was not statistically significant (P=0.40). None had hyperfiltration, defined as a GFR of more than 120 ml/min per 1.73 m2. 4 patients had proteinuria 1-3 g/d, 6 patients had 0.3-0.9 g/d and 8 patients had no proteinuria at baseline i.e. <0.2 g protein/day (or <30mg albumin/day).
Renal function during ERT in Fabry men
The renal function of 15 of the19 men could be studied during 4 years of ERT. Twelve of these 15 men had no or mild proteinuria (<1 gram/day) and were enrolled in the study (Fig. 1). The baseline characteristics for the 12 men are shown in Table 4. Five of the patients had slightly reduced GFR, 63-77 ml/min per 1.73 m2, while 7 patients had normal GFR at baseline.
Figure 1: The design of the study of 4 years ERT on GFR in male Fabry patients.
Twelve of the original 19 men had no or mild proteinuria (<1 gram/day) and were enrolled in the study. * One patient died mors subita at age 54 after 3 years in the study, 2 brothers had to be referred to another medical centre (they died later at age 63 and 62) and one patient had just started treatment.
Table 4: Baseline characterisics in 12 men with Fabry disease with 4 years of ERT
Values are mean±SEM, with ranges in parentheses.
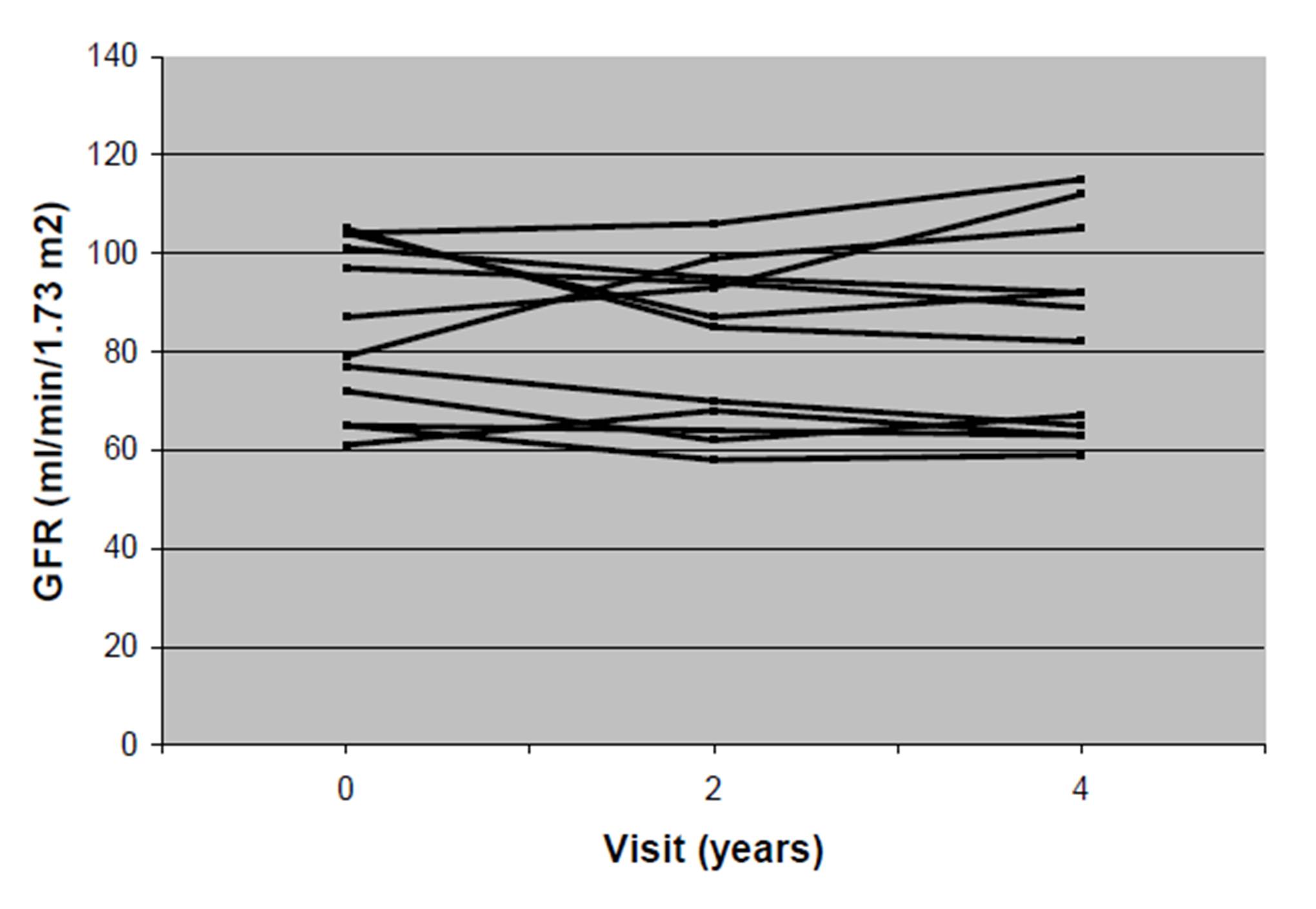
There was no decrease in kidney function, as measured by GFR and serum creatinine, after 4 years of treatment in this group of patients (Table 5). The individual changes in GFR in the 12 men during the 4 years are shown in Fig. 2.
Table 5: GFR and serum creatinine in 12 men with Fabry disease during 4 years of ERT
Values are mean±SEM
Figure 2: Individual changes in GFR (ml/min per 1.73 m2 body surface) in 12 men with Fabry disease during 4 years of ERT
The mild proteinuria remained and was unchanged during the enzyme treatment (data not shown).
Cardiac involvement in Fabry men
Echocardiographic data in the 19 Fabry men at baseline
The cardiac function by echocardiography of 19 of the 19 men could be studied at baseline. Eleven of the 19 male Fabry patients (58%) had LV hypertrophy (age 45.9±2.8, range 30-59 years). It was estimated that 1 patient had severe, 5 had moderate and 5 had mild LV hypertrophy. Eight patients (42%; age 24.4±2.3, range 15-33.5 years) had normal echocardiography. The patients with LV hypertrophy were older than the patients with normal echocardiography (P=0.01).
Echocardiographic data during ERT in Fabry men
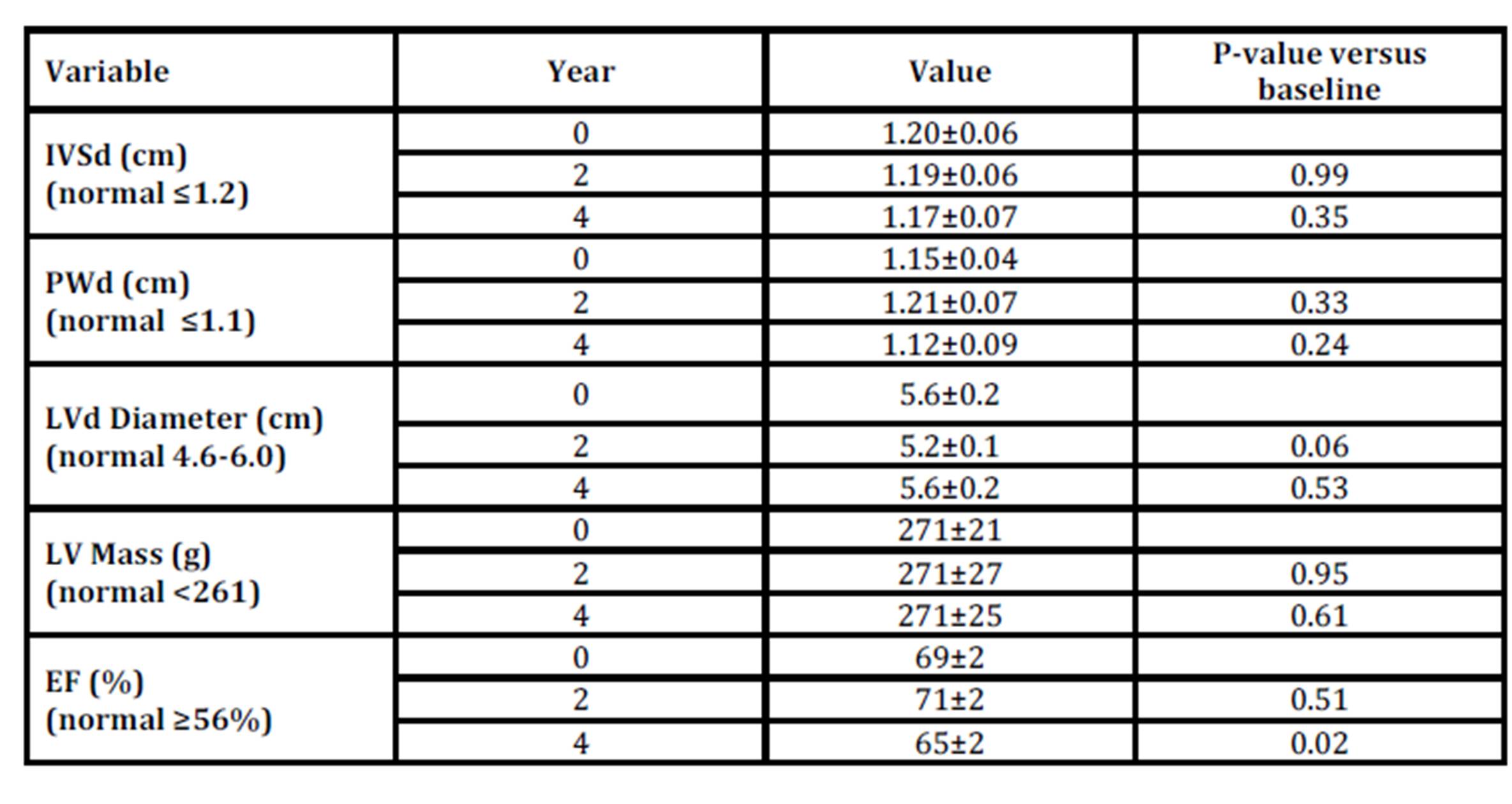
The cardiac function by echocardiography of 10 of the 19 men could be studied during the full study duration of 4 years. Three of these 10 patients had mild and one man had moderate LV hypertrophy. No cardiac deterioration was observed during ERT (Table 6). There was a slight decrease of the EF within the normal range after 4 years (Table 6).
Table 6: Cardiac data in 10 men with Fabry disease during 4 years of ERT
IVSd, inter ventricular septum, diastole; PWd, posterior wall dimension, diastole; LVd, left ventricular, diastole; LV, left ventricular; EF, ejection fraction.
Valvular leakage (regurgitation) was very frequent in the 19 male Fabry patients (data not shown).
Concomitant medication was similar throughout the study except for one patient who received a low dose of an ACE inhibitor and a low dose of a sustained-release preparation of furosemide as well as warfarin during the second half of the study. Three patients had the beta blocker metoprolol in a dose of 50-100 mg, one patient had 75 mg acetylsalicylic acid and one patient had 40 mg furosemide during the study.
Cerebrovascular affection in Fabry men
Fourteen of the original 19 patients could be investigated with an MRI scan of the brain at baseline. Seven patients showed imaging evidence of cerebral involvement (white matter lesions and/or lacunar infarctions) on MRI scans of the brain and 7 patients showed normal MRI scans. The patients with cerebrovascular lesions were older than the patients without lesions, age 50.4±2.4 years (range 40-59) and 28.5±3.1 years (range 15-37.5), respectively (P=0.02).
The cerebrovascular affection by MRI of 11 of the 19 men could be studied after 4 years of ERT. Five patients showed normal MRI scans. Two of the 6 men with signs of previous cerebrovascular lesions showed additional white matter lesions or lacunar infarctions on their MRI scans. These events had been clinically silent. One of the 2 patients had paroxysmal atrial fibrillation and started anticoagulation treatment with Warfarin during the study.
Auditory function in Fabry men
The auditory function was unchanged during the study (data not shown). Gb3 levels in urine and plasma in Fabry men
There was a marked decrease of Gb3 levels in urine and plasma both after 2 and 4 years of ERT compared with baseline, (Table 7).
Table 7: Gb3 levels in urinary sediment and plasma in 15 men with Fabry disease during 4 years of ERT.
Body weight for Fabry men
The body weight for the 15 enzyme treated men increased from 74.5±3.7 kg to 78.7±4.4 kg after 2 years (p=0.01) and remained at this level 78.3±3.4 kg after 4 years of ERT (p=0.05).
Baseline renal function and cardiac involvement in Fabry women
Baseline GFR and proteinuria in Fabry women
Two of the 15 women had reduced GFR combined with proteinuria, 1-3 g/d, at baseline. One of them, aged 53, started ERT due to severe manifestations of her disease. She had endstage renal disease and had started dialysis. Four years after baseline, a kidney transplant was performed. The other woman, aged 39, had GFR 66 ml/min per 1.73 m2 at baseline. She started ERT due to suspicion of decreasing kidney function combined with proteinuria 3 years after the baseline visit.
Thirteen of the 15 women had normal GFR (94.6±5.1 ml/min per 1.73 m2) and no proteinuria at baseline i.e. <0.2 g protein/day (or <30 mg albumin/day). None of the 15 women had hyperfiltration, defined as GFR of more than 120 ml/min per 1.73 m2.
Baseline cardiac involvement in Fabry women
Seven of the 15 women (47%; age 45.7±5.8, range 25.5-64 years) had LV hypertrophy. Of these 7 patients, 6 had mild LV hypertrophy and 1 woman (aged 55) with the cardiac variant of Fabry disease had severe hypertrophic cardio since many years. She had previously received ERT with agalsidase alfa for 2 years at another hospital, but this treatment was terminated due to lack of clinical effect. Three years later she was restarted with ERT (agalsidase beta) at our centre. Eight of the 15 women (53%; age 41.5±3.6, range 21.5-54 years) had normal echocardiography. There was no age difference between those women with normal echocardiography and those with hypertrophy (P=0.61).
A slight valvular leakage (regurgitation) was frequent in the 15 female Fabry patients (data not shown). ERT in the female Fabry patients
During the study period, in total 4 women of the 15 women received ERT at a certain point in time, as a result, 11 of the female Fabry patients were not receiving ERT based on a clinical point of view.
Two of the 15 women started ERT (agalsidase beta) immediately after the baseline visit at ages 58 and 53 years, respectively. The reason for starting this treatment was that the first patient had slight left ventricular hypertrophy as well as acroparesthesia and the second patient had endstage renal disease.
A third woman with only cardiac symptoms had previously been treated with agalsidase alfa for 2 years at another hospital, but this treatment was terminated due to lack of clinical effect. Three years later, at age 55, she restarted ERT (agalsidase beta) at our centre.
Three years after the baseline visit, a fourth woman, aged 42, had to start ERT due to a moderate renal failure combined with proteinuria.
Discussion
With our study we wish to give a survey of the past and present situation concerning Fabry disease in Sweden. We present the baseline characteristics of the patients, symptoms of Fabry disease and the results of ERT. When we started this study very little was known about the Fabry community in Sweden. The care centre for Fabry patients in Sweden was organized by one of the authors (J-OJ) in 2001. Another of the authors (JA) had previously started ERT treatment of the first patients in September 2000. All patients came to our clinic from public healthcare and there was no active search for patients. Up to now we have found 21 different Fabry families but some families might still be unknown to us.
During the first 12-18 months we started ERT with agalsidase alfa (Replagal®) 0.2 mg/kg body weight every other week and thereafter the patients were switched to agalsidase beta (Fabrazyme®) using half of the recommended dose i.e. 0.5 mg/kg body weight every other week, up to 48 months (personal communication J-O Johansson et al., 2003).
Which of the two enzymes that should be used and in what dose has been a topic of discussion.The two products were approved in Europe 2001 with a five-fold difference in dose but a similar pricing. Later studies have shown that the two products have virtually identical biochemical profiles (enzyme activity per mg protein, uptake in fibroblasts, biodistribution and antibody cross-reactivity) (Lee et al., 2003, Desnick 2004, Schaefer et al., 2009). A randomized, open-label study supports the fact that for both agalsidase preparations, a dose of 0.2 mg/kg body weight every other week is suboptimal (Vedder et al., 2007). Thus, a decision about which product to use has been difficult to make on a rational basis.
In one randomized, double-blind, placebo-controlled trial with agalsidase beta 1.0 mg/kg of 82 Fabry patients, the treatment slowed progression to the composite clinical outcome of renal, cardiac and cerebrovascular complications and death compared with placebo in patients with advanced Fabry disease (Banikazemi et al., 2007). Furthermore, an open-label, phase III extension study involving 58 Fabry patients has shown sustained long-term renal stabilization after 41/2 years of treatment with agalsidase beta 1.0 mg/kg. However, despite ERT, some patients showed a progression of renal disease. A prognostic unfavourable sign was the occurrence of high proteinuria (>1 g/24 h) and evidence of sclerotic glomeruli pretreatment (Germain et al., 2007). As a result, patients who do not exhibit evidence of heavy proteinuria and glomerular sclerosis at baseline can be expected to achieve effective stabilization of renal function with agalsidase beta. On the other hand, for patients with a proteinuria exceeding 1 gram/day and a high portion glomerular sclerosis, renal disease progression can be expected despite ERT (Banikazemi et al., 2007, Germain et al., 2007, Schaefer et al., 2009).
In the present study we examined the effect of 4 years of ERT on kidney function in a cohort of male Fabry patients with no or mild proteinuria (<1 gram/day). There was no deterioration of kidney function as measured by GFR and serum creatinine during the treatment. The finding indicates that kidney function has been stable and may have been protected by the given ERT, although the dose of agalsidase beta was half the recommended dose, i.e. 0.5 mg/kg during the last 2.5-year phase of the study. In a recent study by Tøndel et al., renal biopsies were performed at baseline and after 5 years of ERT. Different ERT doses were used and the low dose was 0.2 mg/kg of either agalsidase alfa or beta and the high dose was either 1 mg/kg agalsidase beta every other week or 0.4 mg/kg agalsidase alfa every other week or 0.2 mg/kg agalsidase alfa every week. A highly significant correlation between podocyte Gb3 clearence and cumulative agalsidase dose was seen. No effect on the podocyte inclusion was seen in the low-dose group whereas sustantial clearence was seen in patients with the highest doses of agalsidase. However, irrespective of drug dosage, complete clearence of Gb3 inclusions was seen in the glomerular endothelial cells and mesangial cells in all patients (Tøndel et al., 2013). The clinical significance of podocyte damage in Fabry disease has previously been highlighted (Najafian et al., 2011). Controls without ERT were not included in our study, but the natural history of Fabry patients has been described in some studies. The mean rate of decline in GFR has been between 12.2 (Branton et al., 2008) and 2.93 ml/min per 1.73 m2/year for male Fabry patients (Schiffmann et al., 2009). It is reasonable to believe that the GFR in our group of patients would have decreased during the 4-year study if ERT had not been given. There was a low use of concomitant medication in the study and the indication was cardiac symptoms. It is unlikely that the medication had any effect on the renal outcome of the study.
Between the summer of 2009 and the spring of 2012 there was a global shortage of enzyme for treatment of Fabry disease due to production problems of agalsidase beta (Linthorst et al., 2011). It has previously been suggested that a lower dose of agalsidase beta may be sufficient in some, but not all, Fabry patients (Lubanda et al., 2009). Our study supports the idea that a reduced dose of enzyme may be used during a shorter time. After publication of the study of Banikazemi et al., the dose of agalsidase beta was increased to 1.0 mg/kg (Banikazemi et al., 2007).
In a study of 32 Fabry patients, cardiac morphology and function were studied during 3 years of treatment with agalsidase beta 1.0 mg/kg. In patients without fibrosis, ERT resulted in a significant reduction in left ventricular mass, an improvement in myocardial function and higher exercise capacity. In contrast, patients with mild or severe fibrosis showed a minor reduction in left ventricular hypertrophy and no improvement in myocardial function or exercise capacity (Weidemann et al., 2009). Of the 10 men who were examined by echocardiography, most had mild or no left ventricular hypertrophy. Since it can be assumed that most of these Fabry men had no or very slight myocardial fibrosis, a cardioprotective effect of ERT could be expected. The left ventricular mass was unaltered during the 4 years of ERT, despite the use of a reduced dose of agalsidase beta during the second phase of the study.
As could be expected, the Fabry men with imaging evidence of cerebral involvement were older than the men with normal MRI brain scans. Also, the men with left ventricular hypertrophy were older than the ones with normal left ventricels. These findings are in accordance with the well-known fact that Fabry disease is a progressive disease with increased symptom severity with increasing age. In a recent study Weidemann et al., showed that despite ERT; clinically meaningful events continue to develop in patients with advanced Fabry disease (Weidemann et al., 2013). Thus, in the advanced stages of Fabry disease, specific treatment can decrease symptoms, but not prevent progression towards organ damage.
At the baseline visit, both the men and women showed a similar prevalence of complaints and symptoms compared to an overview of the literature of Fabry patients from other countries (Vedder et al., 2007). However, there were very few renal complications among the Fabry women, although one woman had endstage renal disease. The manifestations among Fabry women seem to range from asymptomatic to as severe as Fabry men (Desnick et al., 2001, Mac Dermot et al., 2001, Patel et al., 2011). The Fabry women should be closely followed and evaluated to monitor the progression of the disease. ERT should be started in case of renal, cardiac or cerebrovascular affection (Mehta et al., 2010). Symptoms of acroparesthesia may also indicate that ERT may be necessary.
The marked decrease of Gb3 levels in urine and plasma is well in accordance with many other studies. One limitation of the study is that the patients have been treated with two different enzyme preparations. Another limitation is the relatively low number of patients. As in any rare genetic disease, it is difficult to enrol large number of patients into clinical studies. This difficulty is even more pronounced when studying Fabry disease, because the heterogenous progression of the disease makes it difficult to obtain a cohort of patients who are at a similar stage of the disease. During the study period, 2 patients also had to be referred to another medical centre which resulted in even fewer patients in the study.
During the last five years only a few new patients have been diagnosed with Fabry disease in Sweden. Misdiagnosis of the disease has been common and the mean delay from onset of symptoms, mostly acroparesteshia, to correct diagnosis was 22.1 years in males and 23.8 years in females (Table 1). The prevalence of Fabry disease in Sweden is estimated at 1 in 187 000 according to our calculation (men and women together). However, our calculation may underestimate the true prevalence of the disease since some Fabry families might still be unknown to us. Today there are two centres for the Fabry patients in Sweden, one in Gothenburg and one in Stockholm.
In conclusion, the 4 years of ERT for men with no or mild proteinuria resulted in a stabilization of kidney function and there was no increase of the left ventricular mass. There was a marked decrease of Gb3 levels in urine and plasma during ERT. Swedish Fabry patients of both genders showed a similar prevalence of classic Fabry symptoms compared to Fabry patients from other countries, although the Swedish Fabry women had less renal complications.
Acknowledgements
The authors thank the Fabry nurses Annica Alklind and Anna-Lena Jönsson for their excellent care of the patients, doctor Anna Bäckström (Bendix) for excellent help with starting this study, doctor Ingrid Ringdahl Rossmann for help with the dermatology evaluations, doctor Hossein Shams for help with the ophtalmological evaluations, the staff at the Department of Audiology for help with the audiometry examinations, the staff at the Institute of Clinical Neuroscience for their excellent technical assistance, Ph.D. Kirsten Rosenberg in Copenhagen and professor Andreas Gal in Hamburg for help with the mutation analyses.
We also thank Gert Frej and his company Penny No One Ltd. for their financial support for three months during the writing of this report. This private sponsor had no involvement in any part of the manuscript.
Disclosure
J-O Johansson has received research funding and speaker’s fees from lectures on the management of Fabry disease in Sweden from Genzyme Corporation. J-E Månsson has received speaker’s fees from lectures on lysosomal storage diseases from TKT-5S (Shire HGT) and Genzyme Corporation.
3. Desnick RJ, Ioannou YA, Eng CM (2001) Alpha-galaktosidase A defiency: Fabry disease. In Scriver CR, Beaudet, AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The metabolic and molecular bases of inherited disease. 8th ed. Vol. 3. New York: McGraw-Hill, 3733-3774.
6. Johansson J-O, Ahlmén J, Månsson JE, Sonne P (2003). Uncomplicated switch from Replagal to Fabrazyme in Fabrypatients. 4th European round table on Fabry disease. Munich, Germany, October 17-18. Expectations of enzyme replacement therapy. Poster 49.
17. Najafian B, Svarstad E, Bostad L et al (2011) Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int 79: 663-670.
18. Nilsson-Ehle P, Grubb A (1994) New markers for the determination of GFR: Iohexol clearance and cystatin C serum concentration. Kidney Int 46, suppl 47: S-17-S19.
26. Vedder AC, Linthorst GE, Houge G et al (2007) Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PloS One Jul 11; 2(7): e598. Google Scholar
28. Waagstein F, Caidahl K, Wallentin I, Bergh CH, Hjalmarson A (1989) Long-term beta-blockade in dilated cardiomyopathy. Effects of short- and long-term metoprolol treatment followed by withdrawal and readministration of metoprolol. Circulation Sep;80(3): 551-563. Google Scholar