Introduction
Kabuki Make up Syndrome(KMS) is a very rare genetic disorder with a prevalence of 1 in 32000 live births in Japanese population¬.1 KMS has five cardinal manifestations which includes characteristic facial features, skeletal abnormalities, dermatoglyphic abnormalities, mild to moderate mental retardation and postnatal growth insufficiency.2 Recently some atypical features like chronic and/or severe diarrhea, diaphragmatic defects, pseudarthrosis of the clavicles, vitiligo, and persistent hypoglycemia, severe autoimmune thrombocytopenia, cerebellar vermis atrophy, and myopathic features have also been described.3,4 KMS has an autosomal dominant inheritance with sporadic mutations. MLL2 gene is the most common gene involved, recently point mutations in the KDM6A gene has also been reported in association with KMS.5,6
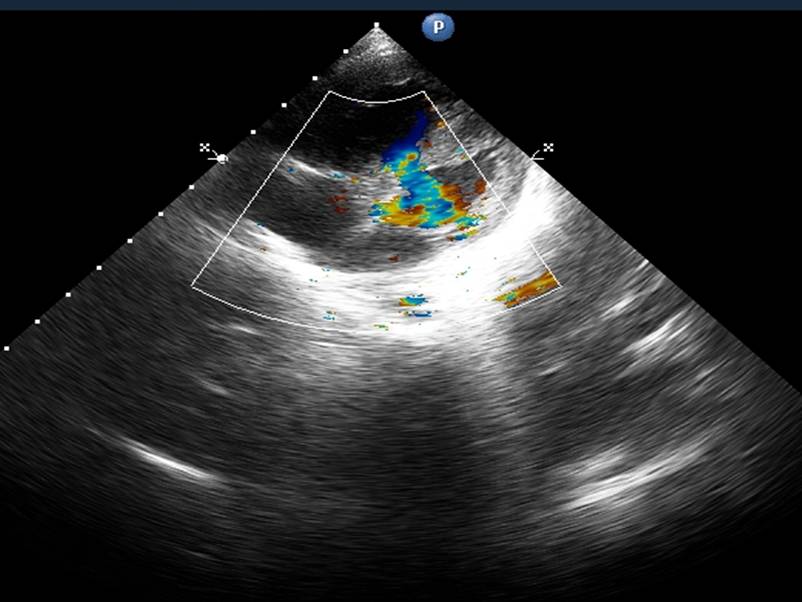
Various congenital heart defects like single ventricle with common atrium, ventricular septal defect, atrial septal defect, tetralogy of Fallot , coarctation of aorta , patent ductus arteriosus, aneurysm of aorta , transposition of great vessels , right bundle branch block, and “parachute” mitral valve, coarctation of aorta, bicuspid aortic valve have been described in KMS since 1980s. The association of congenital heart defects with KMS has been well established, though combination of heart defects in association with this syndrome has been rarely reported. Here we describe a one and half year old Indian (non-Japanese) female child with KMS who had multiple congenital heart defects and ultimately developed eissenmenger’s physiology.
Case Report
One-and-half year old Indian (non-Japanese) female child presented with feeding and breathing difficulties since two months of age. The child was the first child of non-consanguineous parents delivered at home, with no antenatal, natal or post natal complications. The child weighed normally at birth but was floppy (hypotonic) since birth according to the parents. The child was first brought to medical attention at two months of age with infrequent follow-up visits.
The past history was significant for several episodes of respiratory tract infections not associated with cyanosis since two months of age, which according to the parents has improved in the past few months. There was no history suggestive of malabsorption syndrome, seizures, storage disorders, defects of metabolism and chronic infections.
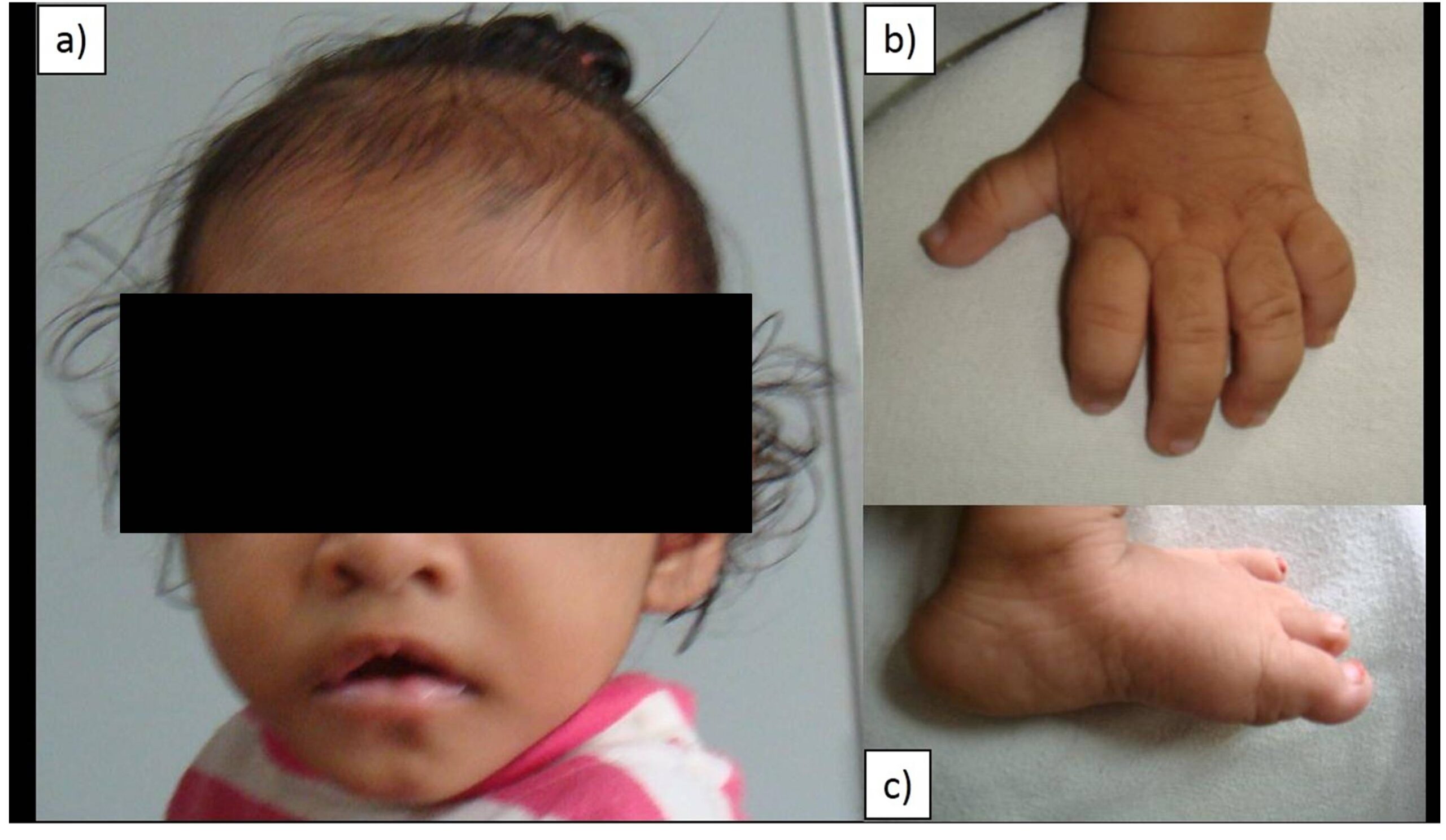
Her weight (6 kg) and her length ( 67 cm) were less than 3rd percentile for age according to World Health Organization(WHO) growth charts. Her head circumference was 42 cm which was less than 3 Standard deviation for age according to World Health Organization (WHO) growth charts; as well her weight for length was between 1 Standard deviation and 2 Standard deviation for age. Thus the child had failure to thrive, severe stunting and microcephaly. On examination, the child had characteristic facial features (Figure 1) — interrupted eyebrows, long palpebral fissure, depressed nasal bridge, low set ears, high arched cleft palate, short fifth digit with clinodactyly and finger pads, hypotonia, overlapping of toes and rocker bottom feet. The child is unusually sociable with a happy disposition and had delayed milestones in all domains.
Skeletal survey revealed spina bifida at T9 and T11 with butterfly vertebra at T10 and T11 and mild scoliosis with concavity toward right. Ultrasonography of abdomen was normal. Karyotyping done on peripheral lymphocytes was normal.
Discussion
Kabuki Make up Syndrome is a multiple congenital anomaly syndrome which was first described in the Japanese population in 1981 .7,8 Some major, minor and behavioral features have been described with this syndrome.9 Major features consists of characteristic facies (100%), long palpebral fissures(99%), abnormal dermatoglyphics(96%), short nasal septum (92%), persistent finger pads(89%), malformed ears(87%), arched eyebrows(85%), Intelligent quotient<80(84%), depressed nasal tip (83%), short 5th digit (79%), joint laxity(74%), high arched palate(72%), abnormal dentition(68%), hypotonia(68%), short stature (55%)and ptosis(50%). Minor features include cardiovascular abnormality(42%), cleft lip/palate(35%), scoliosis(35%),deformed vertebrae or rib(32%), kidney and urinary tract malformation (28%) , premature thelarche (28%), hearing loss(27%), lower lip pits(27%), cryptorchidism (24%), preauricluar pits(22%), hip dislocation(18%) and seizures(17%). Behavioral features include having a happy disposition(87%), liking routine(74%), unusually sociable(50%) and engaged in minimal interaction with others (30%).9 The index child had 7 major(characteristic facies, long palpebral fissures, persistent finger pads, arched eyebrows, short fifth digit, hypotonia, short stature) 4 minor( cardiovascular abnormality, cleft palate, scoliosis, deformed vertebrae) and 2 behavioral features(happy disposition, unusually sociable) mentioned above.
Various congenital heart defects have been described in this syndrome since 1980s. Cardiovascular anomalies has been described in 31-58% of patients with Kabuki syndrome in various studies most of which had described isolated heart defects10,11. Particularly the incidence of cardiovascular anomalies seems to be lower in the Japanese population with one review of 62 Kabuki patients reporting an incidence of 31%.2 While the incidence of cardiovascular anomalies in non Japanese population has been reported from 58-83%.12,13.14 Hughes et al ( 1994), in their study of 20 patients with Kabuki syndrome had an estimated incidence of cardiovascular anomalies of 30% in which 25% had more than one documented cardiac malformation which, in all cases, included an aortic coarctation.10
Although more than about 100 cases of cardiac defects in Kabuki syndrome have been described, combined defects have rarely been described. Hughes et al. 1994, described a combination of coarctation with ventricular septal defect , patent ductus arteriosus and bicuspid aortic valve in 5 of their patients.10 But the recent extensive study and review by Digilio et al. 2001, did not reveal any such combined anomalies.11 Almost all the 60 cases in their study and 70 cases reviewed by them had isolated heart defects. Recently, coarctation of aorta with non obstructing anomalous left pulmonary artery branch (aLPA) from right pulmonary artery (RPA) has been reported in a neonate with Kabuki syndrome.17 Another novel case with Kabuki syndrome diagnosed with hypoplastic left heart syndrome, and right-sided partial anomalous pulmonary venous drainage to the inferior vena cava has been reported.16 The authors chose to report this patient as she had multiple congenital heart defects namely, bicuspid aortic valve, coarctation, ventricular septal defect and patent ductus arteriosus and as the child also developed pulmonary hypertension with reversal of shunt with the ratio of pulmonary flow to systemic flow greater than 1 at a very early age, which has not been reported till date. Although Kabuki syndrome is said to be not associated with serious complications with a good prognosis to adulthood, children with multiple congenital heart defects like ours can have a catastrophic outcome at an early age. This could have been prevented by early diagnosis and appropriate surgical management of the congenital heart defects.
References
1. Malik P, Sharma A, Sakhuja S, Munjal, S, Panda, N. Speech and Language Characteristics in Kabuki Syndrome — A Case Report. The Internet Journal of Allied health Sciences and Practice 2010; 8:1540-580.
2. Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, Ishikawa N et al. Kabuki make-up(Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet 1988;31(3):565-89.
Publisher – Google Scholar
3. Geneviève D, Amiel J, Viot G, Le Merrer M, Sanlaville D, Urtizberea A, et al. Atypical findings in Kabuki syndrome: report of 8 patients in a series of 20 and review of the literature. Am J Med Genet A. 2004 Aug 15;129A(1):64-8.
Publisher – Google Scholar
4. Sethi SK, Faridi MMA. Kabuki Syndrome and Diaphragmatic Defect. Indian Pediatr 2006;43:552-553.
5. Miyake N, Koshimizu E, Okamoto N, Mizuno S, Ogata T, Nagai T, et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med Genet A. 2014 Sep;161(9):2234-43. doi: 10.1002/ajmg.a.36072. Epub 2014 Aug 2.
Publisher – Google Scholar
6. Miyake N, Mizuno S, Okamoto N, Ohashi H, Shiina M, Ogata K ,et al. KDM6A point mutations cause Kabuki syndrome. Hum Mutat. 2014 Jan;34(1):108-10. doi: 10.1002/humu.22229. Epub 2012 Oct 17.
Publisher – Google Scholar
7. Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki-make up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears and postnatal growth deficiency. J Pediatr 1981; 99: 565—569.
8. Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr 1981;99:570-573.
Publisher – Google Scholar
9..Matsumoto N, Niikawa N. Kabuki make-up syndrome; a review. Am J Med Genet. 2003;117C:57-65.
Publisher – Google Scholar
10. Hughes HE, Davies SJ. Coarctation of the aorta in Kabuki syndrome. Archives Dis Child 1994; 70: 512—514.
Publisher – Google Scholar
11. Digilio MC, Marino B, Toscano A, Giannotti A, Dallapiccola B. Congenital heart defects in Kabuki syndrome. Am J Med Genet2001; 100: 269—274.
Publisher – Google Scholar
12. Schrander-Stumpel C, Meinecke P, Wilson G, Gillessen-Kaesbach G, Tinschert S, König R, et al. The Kabuki (Niikawa-Kuroki) syndrome: further delineation of the phenotype in 29 non-Japanese patients. Eur J Pediatr. 1994 Jun;153(6):438-45.
Publisher – Google Scholar
13. Philip N, Meinecke P, David A, Dean J, Ayme S, Clark R, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 16 non-Japanese cases. Clin Dysmorphol. 1992 Apr;1(2):63-77.
Publisher – Google Scholar
14. McMahon CJ, Reardon W. The spectrum of congenital cardiac malformations encountered in six children with Kabuki syndrome. Cardiol Young. 2006 Feb;16(1):30-3.
Publisher – Google Scholar
15. Bhat AH, Davenport J, Cocalis M. Partial anomalous left pulmonary artery along with aortic coarctation in an infant with Kabuki syndrome. Echocardiography. 2012 Jul;29(6):E145-7. doi: 10.1111/j.1540-8175.2011.01651.x. Epub 2012 Feb 13. doi: 10.1007/s00246-008-9197-0. Epub 2008 Feb 19.
Publisher – Google Scholar
16. Shahdadpuri R, Lynch SA, Murchan H, McMahon CJ. A novel constellation of cardiac findings for Kabuki syndrome: hypoplastic left heart syndrome and partial anomalous pulmonary venous drainage. Pediatr Cardiol. 2008 Jul;29(4):820-2. doi: 10.1007/s00246-008-9197-0. Epub 2008 Feb 19.
Publisher – Google Scholar