Introduction
Primary malignant brain tumors are one of the deadliest forms of cancer in both children and adults. Glioblastoma (GBM) has been considered as the most common primary malignant brain tumor in adults that develops de novo or from the progression of lower grade tumor (Phillips et al., 2012). In children GBM is less common. It has a similar histopathophysiology among the various age groups, but is highly heterogeneous with respect to their biological and molecular characteristics. Clinical studies suggest that patients with glioblastoma harbouring, the variant tend to have a worse prognosis (Heimberger et al., 2005; Shinojima et al., 2003).
Almost all current therapies against cancer target differentiated cancer cells, but not cancer stem cells (CSCs), hence CSCs that are more resistant to available therapies and relatively quiescent can escape the treatment that results in recurrence of the tumor. Although normal stem cells and CSCs have many similar properties, the challenges in eradicating CSCs from tumor cells provide a new rationale for CSCs targeted cancer chemotherapy without affecting normal tissue stem cells. Hence finding unique pathway in CSCs is of the utmost importance.
GBMs are heterogeneous group of tumors, consisting of genotypically and phenotypically divergent population of cells (Bigner et al., 1981; Kleihues et al., 1999). As a result, antigenic expression profile shows a significant level of variation among and within the individual GBMs (Wikstrand et al., 1983). Recent evidences suggest that drug transporters may be important for efficient clinical outcome to modulate or block the function of drug transporters in CSCs. It has also been found that human GBMs some drug transporters such as ABC transporters, over express at both mRNA and protein level that correlate with higher risk of death. Therefore, these transporters can be useful for the prognostic predictors as well as designing of new immunotherapeutic strategies for the treatment of malignant gliomas.
Several brain tumor microenvironment proteins such as heparan sulfate proteoglycans (HSPGs) have been found to be involved in the progression of cancer at various stages (Sasisekharan et al., 2002). Heparan sulfate proteoglycans (HSPGs) play vital roles in every step of tumor progression allowing cancer cells to proliferate, escape from immune response, invade neighboring tissues, and metastasize to distal sites away from the primary site. Therefore, finding the significant type of HSPGs in brain tumor microenvironment involved in cancer pathways may provide a better prognosis and will improve the knowledge towards the development of novel therapeutic strategies for the treatment of high grade GBMs.
The modulation of cancer-related miRNAs, either to study their functions or for future applications in therapy, has been receiving a great amount of attention due to several promising results. An important characteristic of miRNA expression modulation is that one miRNA is capable of silencing several genes, in contrast to RNA interference technology, which is capable of silencing only one gene or a few genes belonging to the same gene family. This turns the therapeutic modulation of aberrantly expressed miRNAs into a powerful tool for the treatment of cancer, as well as for the understanding of carcinogenesis.
Gene expression, genomic and proteomic data analysis have showed a great improvement since last few years, but still there are no satisfactory results to discriminate the GBM subtypes. However, in WHO classification for the degree of malignancies shown in the histopathological analysis GBM subtypes have been classified into four grades: Low grade (grade I and grade II) and high grade gliomas (grade III and grade IV) as established by World Health Organization (Louis et al., 2007). Grade I gliomas are relatively benign and identified as best prognostic glioma, whereas grade II tumors consists of some anaplastic cells that further develops into higher grade tumors and shows poor prognosis. Grade III tumors are highly anaplastic and mitotic in nature that becomes fatal even more rapidly as compared to grade I and grade II gliomas. Grade IV gliomas are highly anaplastic and malignant in nature that has been considered as the most aggressive and almost fatal because of its resistance to currently available radio and chemotherapy.
Discrimination of current subtypes of human gliomas on the basis of molecular prognosis will likely to be played important role in developing improved therapeutic strategies. Therefore, there is a great need for understanding the factors involved in tumorigenesis in different tumor subtypes. In this review we will focus on the genes present in the brain tumor microenvironment, drug transporters and miRNA as major target for the development of potential therapies for the treatment of human gliomas.

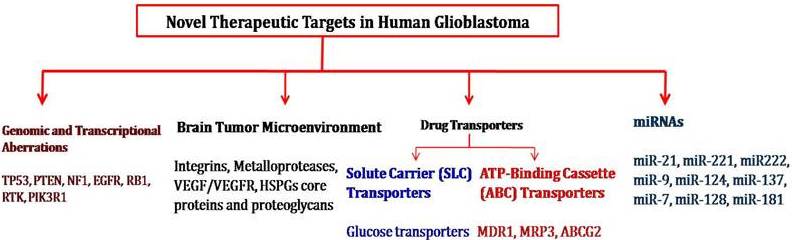
Fig. 1: Novel Therapeutic Targets for the Development of Human GBM Treatment Strategies
Current Progress in Molecular Targeted Therapy and Prognosis for Malignant Glioma
Currently, the examinations used in clinical diagnosis for glioma include computed tomography (CT) scan, magnetic resonance imaging (MRI) scan (Towner et al., 2011), angiogram, Mosaic Analysis (Liu et al., 2011), skull x-ray, spinal tap, myelogram and biopsy. But it is still difficult to diagnose in the earlier period of gliomas, until if we can find some specific biomarkers associated with the early development of gliomas, the diagnosis will be easy to decide. Diagnostic biomarkers are used to detect and identify a given type of cancer in an individual. These markers are expected to have high specificity and sensitivity (Kulasingam et al., 2008).
The wonderful progress in advanced molecular technologies has provided a tremendous amount of data that has altered the way in which we classify and categorize human glioma. From the initial identification of chromosomal aberrations by karyotyping and comparative genomic hybridization, we have rapidly moved to expression array studies and to integrative genomic approaches which have allowed the stratification of several human brain tumors into molecular subgroups (Claudia et al., 2013; Kuan et al., 2010; Gerstner et al., 2009; Hormigo et al., 2011; Mischel et al., 2003). These data have not only increased our understanding of the molecular pathogenesis of human brain tumors, but have also identified prognostic markers and opened new avenues for targeted therapies.
There are two main trends in glioma research that have already yielded and probably will yield further molecular biomarkers of clinical impact: the use of large-scale profiling techniques and, mainly driven by the need to work cost-effectively and to increase sample numbers, the formation of large research networks such as The Cancer Genome Atlas Research Network (TCGA). The Cancer Genome Atlas (TCGA) Research Network has been established to generate the comprehensive catalog of genomic abnormalities driving tumorigenesis, but still only a few molecular factors show promise for prognosis such as methylation status of the MGMT promoter (Hegi et al., 2005).
In an attempt to better understand GBM, many groups have turned to high-dimensional profiling studies such as copy number alterations (Beroukhim et al., 2007; Ruano et al., 2006) and gene expression profiling studies identifying gene signatures associated with EGFR, PDGFRA, IDH1 and NF1 genes expression, clinical features, and survival (Shai et al., 2003; Liang et al., 2005; Phillips et al., 2006; Tso et al., 2006).
In the near future, novel insights into the pathogenesis of gliomas are to be expected from ongoing, large-scale collaborative profiling studies addressing the complexity of genetic, epigenetic, transcriptomic, and miRNA changes by high-resolution array-based techniques or deep sequencing approaches. It appears very likely that these studies will uncover novel molecular markers that may further refine the diagnostic assessment of gliomas. However, the future role of molecular diagnostics in neurooncology, in particular concerning the value of predictive markers will also depend on the development and availability of novel therapeutic alternatives to allow for more sophisticated patient-tailored treatment choices based on molecular profiles.
Genomic and Transcriptional Aberrations Involved in Malignant Glioma
Human cancer cells typically harbour multiple chromosomal aberrations, nucleotide substitutions and epigenetic modifications that drive malignant transformation. The Cancer Genome Atlas (TCGA) has assessed the value of large-scale multi-dimensional analysis of these molecular characteristics in human cancer.
The glioblastoma cancer genome was the first to be characterised in the concerted efforts of the Cancer Genome Atlas project (TCGA) (McLendon et al., 2008). The pilot project screened a total of 587 samples down to 206, which were used to conduct genome-wide analysis of DNA copy number and gene expression, and DNA methylation screening on a total of 2,305 assayed genes. Of the 206 chosen biospecimens, 21 were post-treatment glioblastoma cases and the remaining 185 represented predominantly primary glioblastomas. In the study, upon statistical analysis of mutation significance in the 91 matched glioblastoma-normal pairs selected for detection of somatic mutations in 601 selected genes, eight genes were found to be significantly mutated: TP53, PTEN, NF1, EGFR, ERBB2, RB1, PIK3R1 and PIK3CA.
Two decades of molecular studies have identified important genetic events in human glioblastomas, including the following: (1) dysregulation of growth factor signalling via amplification and mutational activation of receptor tyrosine kinase (RTK) genes; (2) activation of the phosphatidylinositol-3-OH kinase (PI(3)K) pathway; and (3) inactivation of the p53 and retinoblastoma tumour suppressor pathways 1. Recent genome-wide profiling studies have also shown remarkable genomic heterogeneity among glioblastoma and the existence of molecular subclasses within glioblastoma that may, when fully defined, allow stratification of treatment. Albeit fragmentary, such baseline knowledge of glioblastoma genetics sets the stage to explore whether novel insights can be gained from a more systematic examination of the glioblastoma genome.
Brain Tumor Microenvironment Genes Involved in Tumorigenesis
Currently the prognosis for patients with GBM is challenging, with recent advances in imaging, genome sequencing and proteomic approaches, there is great hope for the early diagnosis and treatment of different grades of glioma in more specific line of attack. Large-scale analysis of bulk tumors have revealed significant differences in expression of genes involved in tumor microenvironment such as proteoglycans and immune response related genes (Phillips et al., 2012).
Several signaling pathway genes have been the target of GBM treatment but only limited clinical success has been achieved. Therefore, there is great need to develop the effective therapeutic strategies targeting more comprehensive signaling pathways including modulation by its microenvironment such as many aspects of tumor growth and angiogenesis is regulated by RTK signaling pathway genes, extracellular matrix components (Integrins and metalloproteases, BCL2L12 and VEGF/VEGFR).
Invasion of tumor cells into the healthy brain tissue is facilitated by expression of different proteolytic enzymes like matrix metalloproteinases (MMPs), a family of zinc-dependent endopeptidases (Fillmore et al., 2001; Velasco et al., 2000). They mediate the degradation of protein components of the extracellular matrix (Egeblad et al., 2002). To date, 23 members of the human MMP gene family are known (Nagase et al., 2006). Elevated levels of several MMPs, like for example MMP-1, -2, -7, -9, -11, -12, -14, -15, -19, -24 and -25 have been reported in malignant glioma samples from patients (Velasco et al., 2000; Hur et al., 2000; Lampert et al., 1998; Liano et al., 1999; Nakada et al., 1999; Nakagawa et al., 1994; Nakano et al., 1995), suggesting that their expression is closely related to malignant progression in vivo.
Other brain microenvironment proteins such as Integrins constitute a large family of heterodimeric transmembrane glycoprotein receptors that mediate cell adhesion to a wide variety of extracellular matrix (ECM) proteins. The expression of many integrins (av, a2, a3, a5 and a6, b1, b3, b4, a2b1, a4b1, avb3 and avb5) is reportedly increased in malignant gliomas, whereas expression of a6b1 is decreased relative to normal brain tissue. Clinical investigations of integrin inhibitors in gliomas have focused on the utility of the cyclic RGD-containing peptide cilengitide. Cilengitide (EMD 121974, Merck KGaA, and Darmstadt, Germany) is a cyclic pentapeptide (Arg-Gly-Asp-DPhe-(NMeVal)) that binds to integrins avb3 and avb5 with high affinity (Dechantsreiter et al., 1999; Xiong et al., 2002) and has potent anti-angiogenic and antiproliferative effects in cellular and animal glioma models (MacDonald et al., 2001; Mikkelsen et al., 2008). The initial phase I clinical trials were conducted in both children and adults with recurrent malignant glioma. Cilengitide was well-tolerated, with limited side effects over a broad range of dose levels, and sporadic tumor responses were noted in this largely heavily pretreated patient population (Nabors et al., 2007; MacDonald et al., 2008).
Interaction between the tumor and constituents of its microenvironment are known to regulate malignancy, specifically Heparan Sulphate Proteoglycans (HSPGs) has been shown of much importance as they bind with diverse type of extracellular proteins, including various growth factors and cell adhesion molecules regulating the activity of several ligand-mediated signaling pathways (Smith et al., 2009; Feyzi et al., 1997; Ono et al., 1999; Kreuger et al., 2001; Ashikari-Hada et al., 2004). HSPGs are the component of extracellular environment in normal brain as well as GBM and are present on cell surface and extracellular microenvironment. HSPGs also promote receptor signaling such as with FGF-2 and VEGF by acting as co-receptors (Dhoot et al., 2001; Ai et al., 2003; Gallagher et al., 2001). HSPG-mediated signaling is also critical for normal brain development (Sarrazin et al., 2011). In GBM, expression levels of multiple HSPGs core proteins and modifying enzymes are significantly altered to normal brain.
Targeting these pathway genes and extracellular matrix components especially HSPGs, and related components of the tumor microenvironment has the potential to simultaneously inhibit multiple oncogenic signaling pathways in tumor cells and to disrupt critical tumor-microenvironment interactions. Therefore, identification of the relevant tumor microenvironment interactions will help to effectively target and treat the GBM.
Hspgs Core Proteins and Modifying Enzymes as Target for the Treatment of GBM
HSPGs core proteins are important determinants of cell signaling molecules in tumorigenesis. They specifies localization of proteoglycans and influence both intracellular and extracellular signaling (Rapraeger et al., 2000). For example, the syndecans (SDCs) can serve as a substrate in cell signaling pathways while binds with cytoskeleton proteins with the help of cytoplasmic domains (Whiteford et al., 2011). Manipulation of syndecan-1 expression has been shown to alter the HGF-Met signaling (Ramani et al., 2011) and Wnt signaling in cancer (Alexander et al., 2000). Other HSPGs, such as perlecan (HSPG2) are also found within the extracellular matrix can facilitate the resolution of inflammation (Hayashida et al., 2009). High levels of syndecan-1 in multiple myeloma correlates with poor prognosis and have been associated with increased tumor growth in animal models (Seidal et al., 2000; Yang et al., 2002).
Alteration in HSPGs vary across the tumor subtypes suggesting that there may be subtype specific HSPGs functions in GBM such as proneural GBM subtype is characterized by alteration in platelet derived growth factor receptor (PDGFR) signaling has high SULF-2 expression (Philips et al., 2006).
Understanding the function of HSPGs core proteins and modifying enzymes will be important for the optimization of future therapeutic strategies in context with subtype specific brain tumors (Wade et al., 2013). And can represent significant molecular target to represent clinically relevant, druggable therapeutic target in the treatment of GBM.
Heparan Sulfate Proteoglycans and Extracellular Sulfatases
HSPGs consist of a protein core and heparan sulfate (HS) chain. HSPGs play crucial role in cell signaling pathways; hence, HS chain of HSPGs undergo extensive post translational modifications, including sulfation on the 6-O-position of Glucosamine which is critical determinant for the binding of growth factors and normal development (Bink et al., 2003; Bulow et al., 2004; Esko et al., 2001; Habuchi et al., 2004). Recently discovered extracellular sulfatases, SULF-1 and SULF-2, can activate multiple key signaling pathways (Wnt, Shh, GDNF and PDGF) by removing 6-O-sulfates on HS chains and mobilizing protein ligands from HSPG sequestration in the extracellular microenvironment (Phillips et al., 2012).
The identity and levels of HSPGs core proteins are also important determinants of cell signaling. They also localize specific proteoglycans and influence both the extracellular and intracellular signaling such as syndecans (SDCs) are crucial for cell adhesion and migration. Changes in SDCs expression has been shown to alter the HGF-Met signaling (Darksen et al., 2002; Ramani et al., 2011). Other core proteins such as glypicans (GPCs), agrin (AGRN), serglycin (SRGN) and perlecan (HSPG2) are thought to be involved in tumorigenesis and need to be analyzed in different grades of gliomas to identify the better target.
Since HSPGs regulate multiple upstream signaling pathways critical in malignancies, it is of great interest that a recent class of compound has been developed to inhibit some of these oncogenic functions. Heparan sulfate (HS) mimetics are highly sulphated oligosaccharides that inhibit heparanase, sequester HSPGs- binding factors and SULF-2. Hence, can be considered as more valuable therapeutic molecule to inhibit the oncogenic function and terminate the tumor invasion. While HS mimetics have not been yet tested in GBM, represent a promising strategy.
HSPG are also targets of an endoglycosidase known as Heparanase (HSPE) and generates biologically active fragments of Heparan Sulphae (HS) chain. It is upregulated in many cancers including GBM and has been implicated in increased tumorigenesis, tumor angiogenesis and invasiveness (Hong et al., 2010; Ridgway et al., 2011; Kurokawa et al., 2003).
Various studies suggest that tumors enzymatically modify components of the brain tumor microenvironment and help to drive oncogenic signaling and invasion. Disruption of this partnership may be an important therapeutic strategy for the treatment of GBM and other tumors.
Drug Transporters as Target for Cancer Chemotherapy
Drug transporters are key players in the uptake of nutrients such as sugars, amino acids, nucleosides and inorganic ions into an efflux of the xenobiotic toxic substances out of the cells to sustain cell survival. They have been classified into two major families:
- Solute carrier (SLC) transporters
- ATP-binding cassette (ABC) transporters
Transporter proteins that affect drug absorption, distribution and excretion are called drug transporters and confer resistance/sensitivity to anticancer drugs. The functional characterization of these transporter proteins may play a crucial role in the diagnosis of GBM subtypes. A number of studies have revealed that many transporter proteins are being upregulated in malignant tumors compared to normal tissues, suggesting that a differential expression pattern of their existence in cancer can be exploited as a promising strategy for drug designing, and delivery in various types of tumors including GBM.
Glucose Transporters as a Diagnostic Marker and Correlation with Brain Tumors
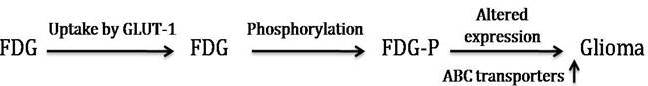
Glucose is the main source of energy in brain tumors (Allen et al., 1972). Its metabolism begins with transport from the serum to cells and continues through the process of phosphorylation catalyzed by hexokinase (HK). Positron emission tomography with FDG is based on the fact that FDG is similar to glucose, is transported across the blood brain barrier (BBB) and cell membranes and then is phosphorylated by HK to FD-G6-phosphate (FDG6P), which accumulates in tissues at a rate proportional to the rate of glucose utilization (Fig. 2). FDG6P is not metabolized further along the glucose metabolic pathways but is slowly dephosphorylated. FDG and glucose differ in their rates of transport and phosphorylation and respective volumes of distribution in brain or tumor tissue. As a result, FDG metabolism is thought to be proportional to, but not quantitatively equal to glucose metabolism in brain tumors (Spence et al., 1998). Glucose transporter (GLUT-1) has been considered as over expressed in various malignant tumors due to increase in rate of glucose uptake and its metabolism (Barker et al., 1997). Positron Emission Tomography (PET) is an advanced imaging tool that measures regional tissue glucose metabolism has gained considerable attention in clinical interest, reporting one of the most important pathophysiological mechanisms for detecting human malignant tumors.
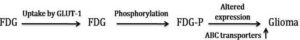
Fig. 2: FDG Metabolism and Gene Expression in Glioma
In brain tumors, it is still to find that GLUT-1 expression pattern in different grades of glioma and various other tumors including lung, lymphoma, melanoma and breast cancer etc., suggesting that identification of GLUT-1 can be a useful diagnostic tool for human gliomas and should be compared to standard physiological characteristics.
Targeting ABC Transporters in Human Brain Tumors
Beyond playing role in drug resistance, ABC transporters are also thought to be involved in tumorigenesis by creating inflammatory microenvironment through transportation of pro-tumorigenic factors (e.g. prostaglandin, cyclic nucleotides and platelet-activating factors etc.) out of cancer cells (Borst et al., 2002; Eilers et al., 2008; Jansen et al., 2003; Sarkadi et al., 2006).
Given the drawbacks of direct transporter inhibition, recent research has focused on elucidating the intracellular signaling pathways that control ABC efflux transporters at the blood brain barrier (BBB). The rationale for this approach is that finding the molecular switches of these transporters will allow selective modulation of transporter function and/or expression for therapeutic purposes in different clinical scenarios. Such an approach has three advantages. First, although direct transporter inhibition leaves little control over the extent and duration of barrier opening, targeting transporter regulation allows more subtle changes in transporter activity so transporters can be turned off for brief, controlled periods of time. Exploring this approach provides a time window during which the barrier is open to deliver normally nonpenetrating CNS drugs and reduces the risk of harmful toxicants entering the brain. Second, direct inhibition that blocks transporter activity can only be used to improve brain drug delivery. Targeting transporter regulation, on the other hand, can also be used specifically to increase transporter expression and/or activity to increase barrier function for therapeutic purposes. Such a strategy provides the opportunity to enhance brain protection and minimize central side effects during treatment of peripheral diseases. Third, BBB ABC efflux transporters are affected by and contribute to CNS disease pathology (Miller et al., 2008; Shen et al., 2010).
Studies show that the efflux transporters P-glycoprotein, BCRP, and MRPs are involved in CNS disorders such as epilepsy, brain cancer, and AD where their role extends beyond that of solely extruding drugs. In such cases, direct transporter inhibition will not be of therapeutic benefit, whereas targeting the signaling pathways that control these transporters could be a useful therapeutic strategy. Thus, understanding the signaling pathways through which BBB ABC efflux transporters are regulated provides opportunities to protect the brain during treatment of peripheral diseases, to improve brain drug delivery to treat CNS disorders, and to prevent pathogenesis or slow the progression of CNS diseases.
The release of pro-inflammatory cytokines in these CNS diseases triggers profound changes in gene expression in the brain and the BBB including changes in expression of ABC drug efflux transporters. ABC transporters might help to create inflammatory microenvironment by transporting pro-tumorigenic factors such as prostaglandins, leukotrienes, cyclic nucleotides, and platelet-activating factor out of cancer cells. Once in the extracellular fluid, these molecules are thought to bind to their extracellular G protein–coupled receptors via autocrine or paracrine signaling and sustain cancer-related inflammation (Fletcher et al., 2010). This mechanism is not yet fully understood, and the exact role ABC transporters play in tumorigenesis needs to be elucidated. However, based on observations that ABC transporters are highly expressed in tumors and tumor stem cells, that they potently confer chemotherapy resistance, and that they may be involved in tumor generation, proliferation, and survival, these transporters may be major targets in cancer therapy (Anika et al., 2010). Thus, identifying pathways that can be specifically targeted to decrease the expression of ABC transporters in cancer will be an important step toward improving chemotherapy of cancer, especially brain tumors.
Pathway Inhibition: Emerging Molecular Targets for Treating Glioblastoma
The current standard of care for newly diagnosed glioblastoma is surgical resection with concomitant daily Temozolomide (TMZ) and radiotherapy followed by TMZ (Stupp et al., 2005). However, almost all patients with glioblastoma experience disease recurrence. Blocking the activation of oncogenic pathways, either at ligand-receptor interaction level or by inhibiting downstream signal transduction pathways, inhibits the growth and proliferation of tumor (Toschi et al., 2008; Hagerstrand et al., 2006; Du et al., 2009; Rieger et al., 2008; Allen et al., 1999).
Within the heavily researched pathways, the overarching theme in glioblastoma therapy is that monotherapies demonstrate limited efficacy. Because single-agent therapies have shown no significant benefit, it is critical to begin designing rational combinations. Different RTK inhibitors combined with PI3K/mTOR dual inhibitors or antiangiogenic agents combined with Akt inhibition are already being examined.
Several studies have been demonstrated various novel agents that inhibit targets identified by screening methods or that are based on preclinical studies and experience in other tumors (Perry et al., 2012; Wachsberger et al., 2012). However, further analyses of clinical and molecular data derived from the trials are necessary to verify the relevance of these targets to glioblastoma.
Micro RNA and Gliomas
Micro RNA is small non-coding RNA molecules (~20-25nt), that regulates gene expression at post-transcription level in a sequence specific manner. A number of miRNA has been reported to be overexperessed in GBM (Conti et al., 2009). In a recent report, it has been found that seven miRNAs expression pattern (miR-21, miR-128, miR-132, miR-134, miR-155, miR-210 and miR-409) allows to discriminate between oligodendroglioma and glioblastoma (Conti et al., 2009; Ciafre et al., 2005; Lages et al., 2011). They are thought to play very important role in cell cycle control, cell proliferation, differentiation and apoptosis (Esquela-Kerscher et al., 2006). Therefore, it can provide a considerable prognostic biomarker and therapeutic target in glioma and other cancer types. To reach this goal different micro RNA expression levels should be identified in comparison with surrounding of the normal brain.
However, the alteration of miRNA expression profile in glioma has been also found to be associated with neoplastic agents (Slaby et al., 2010; Ujifuku et al., 2010; Corsten et al., 2007), thus, functional characterization of these aberrantly expressed microRNAs indicates that they might also function as oncogenes and tumor suppressors (collectively named as “oncomiRs”) may provide potential diagnostic and prognostic tumor biomarkers and represent new therapeutic targets for cancer therapy (Esquela et al., 2006).
Micro RNAs Biogenesis
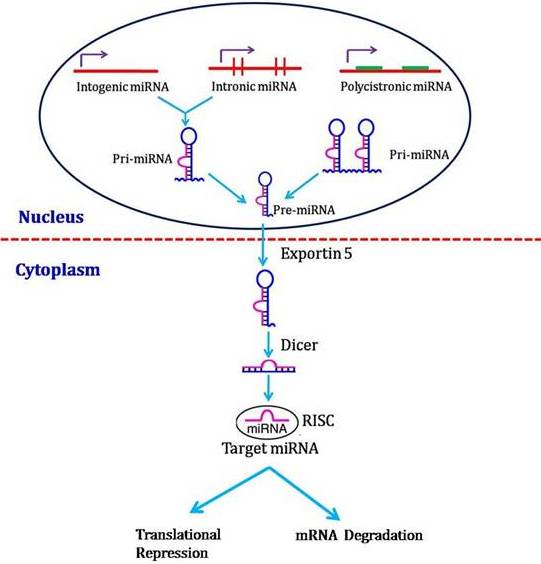
miRNAs biosynthesis involves a transcription of hairpin-shaped long transcripts generated by RNA polymerase II, followed by the endonucleolytic cleavage, mediated by two type III ribonucleases enzymes (RNAse III) known as Drosha (in nuclei) and Dicer (in cytoplasm). miRNA biogenesis is controlled by the multistep miRNA processing pathway (Winter et al., 2009). It is located in intronic regions or within exons of protein-coding genes that can be cleaved by splicing machinery to generate the pre-miRNA (Ruby et al., 2007). This nuclear processing is followed by the transport of pre-miRNA from the nucleus into the cytoplasm via exportin-5 (XPO5) (Yi et al., 2003). In the cytoplasm, the pre-miRNA is cleaved into an approximately 19–25-nt-long mature form of the miRNA by the ribonuclease Dicer (DICER1) (Bernstein et al., 2001) or by a Dicer-independent maturation process that is beginning to be revealed (Cifuentes et al., 2010). The mature miRNA is loaded into the RNA-induced silencing complex (RISC), where it initiates translational repression or degradation of target mRNAs (Sevignani et al., 2006; Zhang et al., 2007). Thus, miRNA biogenesis is tightly controlled by a set of proteincoding genes that eventually lead to the production of functionally mature miRNAs in the cytoplasm.
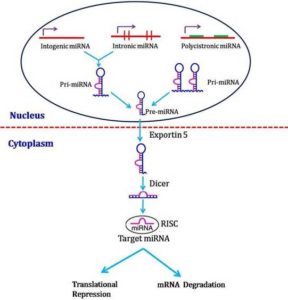
Fig. 3: miRNA Biogenesis
Involvement of Mirnas as Novel Biomarkers in Malignant Gliomas
Biomarkers are major indicators that serve for diagnosis, prognosis and medication. At present, miRNAs are emerging as novel and practical biomarkers in oncology due to their surprising stability, to be detected in fresh and frozen tissues, but also in body fluids and even formalin-fixed paraffin-embedded samples. Recent studies have raised a possibility that miRNAs may function as predictive markers for therapeutics.
Temozolomide (TMZ) is a well-known alkylating agent against gliomas, which methylates guanines in the O-6 position. Accumulating O-6 methyl guanines lead to DNA mismatch during replication and ultimately result in cell death. Although MGMT (methylguanine methyl transferase) promoter methylation has been proved to be a strongest marker to predict the TMZ response in low-grade and high-grade gliomas (Hegi et al., 2005; Everhard et al., 2006; Stupp et al., 2009), it may not be the only indicator. Recent research indicates that miR-181b/c, miR-195, miR-455-3p and miR-10a-3p, rather than MGMT promoter methylation, predict response to concomitant chemoradiotherapy and acquired TMZ resistance in GBM cell line and patients (Ujifuku et al., 2010; Slaby et al., 2010). In addition, increasing evidences indicate that miRNAs also contribute to other drug response. Various highly efficient MGMT inhibitors have been developed for sensitizing tumor to TMZ (McElhinney et al., 2003). The emerging roles of miRNAs in drug resistance of TMZ and other antineoplastic agents give us a novel angle to include miRNAs as therapeutic targets.
A number of studies have reported changes in the steady-state levels of mature miRNAs in glioma (Ciafre et al., 2005; Kim et al., 2011; Sana et al., 2011). One of the best-characterized events is the elevation of oncomiRs miR-21 and miR-221 (Sana et al., 2011; Chan et al., 2005; Conti et al., 2009; Papagiannakopoulos et al., 2008; Zhang et al., 2010; Moore et al., 2010). However, the mechanisms underlying deregulation of miRNA biogenesis in glioma are unknown. The lack of correlation between primary miRNA (primiRNA) transcripts and mature miRNAs in tumours and the association of miRNA processing factors with tumourigenesis in cell culture and mouse model studies indicate that deregulation of the biogenesis pathway is likely to be a key player in the aberrant miRNA expression profiles observed in cancer.
Table1: Major miRNAs Implicated in Glioma 
miRNAs: A Future Scenario
There is a desperate need for developing innovative therapies for GBM. The clinical potential of delivering tumor-suppressive miRNAs or inhibitors of oncogenic miRNAs has been recently advocated (Brown et al., 2007; Edge et al., 2008; Gomez-Manzano & Fueyo, 2010; Wu et al., 2009). The ability of individual miRNAs to target multiple genes/pathways and, at the same time, the capacity of multiple miRNAs in addressing the same 3’ UTR of a single gene, could be a major advantage, especially given the therapeutic necessity of simultaneously targeting multiple pathways in a multifaceted disease such as GBM.
More interestingly, with recent advances in detecting and quantifying miRNAs in tissue, serum, and cerebrospinal fluid it appears increasingly likely that they would be clinically useful as specific and reliable biomarkers patterns for brain tumors and other cancer diseases (De Smaele et al., 2010).
Conclusion
As discussed above, the interest for HSPGs, ABC transporters and miRNA as innovative approach to target these molecules in the treatment of GBM is rapidly increasing. However, as with any novel therapeutic tool further studies are required for their effective translation into clinics in concern with glioma specific tumor targeting.
It is likely that many of the novel therapies discussed in this work will demonstrate greater efficacy when paired with the more studied targeted therapies. Because many of these targets are within the same signaling cascade, inhibiting pathways horizontally rather than vertically should remove some of the compensatory mechanisms glioblastomas use to overcome treatment. It is also important to note that many of the molecular biology advancements will be augmented by advancements in current treatments. Thus, identifying the exact pathway targeted to decrease the expression of ABC transporters and other tumor transcription factors will provide and important target towards the development of novel chemotherapy in the treatment of brain tumors.
As it has been proposed that HSPGs play critical role in tumor growth and metastasis, makes them ripe target for new therapeutic strategies to block heparan sulfate expression or function in vivo clearly need to be investigated. Refining our understanding about the structural and functional alterations within the tumor microenvironment will lead to develop new exciting therapeutic opportunities for the treatment of GBM.
Over the past few years, treatment of brain tumors has shifted towards the designer drugs. Therefore identification of molecular/genetic profiles of tumors and correlative biomarkers of response or resistance to target therapies is of critical importance in order to develop patient specific individual drugs. Because the number of primary brain tumor patients are limited, collaborative efforts will lead to expedited, efficient and rational clinical trial evaluation of new therapeutic targets in GBM prognosis and treatment.
In conclusion, the uncovering of miRNAs has added a new level of complexity to our understanding in genetics of tumors. At the same time, it revealed a new category of therapeutic targets. There are huge amounts of data from researches that are still to be systematized, and new findings on miRNAs involved in GBM are expected from further analysis. And there is need to for better knowledge of miRNA functions, their interrelationships with other cellular processes and the already available treatments against unwanted proliferation can generate new approaches, such as combined therapies, in which the manipulation of miRNA expression can play a pivotal role.
Acknowledgement:
None
Conflict of Interest:
None
(adsbygoogle = window.adsbygoogle || []).push({});
References
Ai, X., Do, A. T., Lozynska, O., Kusche-Gullberg, M., Lindahl, U. & Emerson, C. P. (2003). “QSulf1 Remodels the 6-O Sulfation States of Cell Surface Heparan Sulfate Proteoglycans to Promote Wnt Signaling,” The Journal of Cell Biology, 162, (2) 341-351.
Publisher – Google Scholar
Alexander, C. M., Reichsman, F., Hinkes, M. T., Lincecum, J., Becker, K. A., Cumberledge, S., et al. (2000). “Syndecan-1 is required for Wnt-1 Induced Mammary Tumorigenesis in Mice,” Nature Genetics, 25, (3) 329-332.
Publisher – Google Scholar
Allen, J. D., Brinkhuis, R. F., Wijnholds, J. & Schinkel, A. H. (1999). “The Mouse BCRP1/MXR/ABCP Gene: Amplification and Overexpression in Cell Lines Selected for Resistance to Topotecan, Mitoxantrone, or Doxorubicin,” Cancer Research, 59, 4237-4241.
Publisher – Google Scholar
Allen, N. (1972). ‘Respiration and Oxidative Metabolism of Brain Tumors,’ In: Kirsch WM, Grossi-Paoletti E, Paoletti P, Eds. The Experimental Biology of Brain Tumors, 243-274.
Google Scholar
Ashikari-Hada, S., Habuchi, H., Kariya, Y., Itoh, N., Reddi, A. H. & Kimata, K. (2004). “Characterization of Growth Factor-Binding Structures in Heparin/Heparan Sulfate Using an Octasaccharide Library,” The Journal of Biological Chemistry, 279, (13) 12346- 12354.
Publisher – Google Scholar
Barker, F. G., Chang, S. M., Valk, P. E., et al. (1997). “18-Fluorodeoxyglucose Uptake and Survival of Patients with Suspected Recurrent Malignant Glioma,” Cancer, 79, 115-126.
Publisher – Google Scholar
Beroukhim, R., Getz, G., Nghiemphu, L., Barretina, J., Hsueh, T., Linhart, D., et al. (2007). “Assessing the Significance of Chromosomal Aberrations in Cancer: Methodology and Application to Glioma,” Proceedings of the National Academy of Sciences of the United States of America, 104, 20007–20012.
Publisher – Google Scholar
Bigner, D. D., Bigner, S. H., Ponten, J., Westermark, B., Mahaley, M. S., Ruoslahti, E., et al. (1981). “Hetergeneity of Genotypic and Phenotypic Characteristics of Fifteen Permanent Cell Lines Derived from Human Gliomas,” Journal of Neuropathology & Experimental Neurology, 40, 201-229.
Publisher – Google Scholar
Bink, R. J., Habuchi, H., Lele, Z., Dolk, E., Joore, J., Rauch, G. J., Geisler, R., Wilson, S. W., Hertog, J., Kimata, K. & Zivkovic, D. (2003). “Heparan Sulfate 6-O-Sulfotransferase is Essential for Muscle Development in Zebrafish,” The Journal of Biological Chemistry, 278, (33) 31118–31127.
Publisher – Google Scholar
Borst, P. & Elferink, R. O. (2002). “Mammalian ABC Transporters in Health and Disease,” Annual Review of Biochemistry, 71, 537-592.
Publisher – Google Scholar
Bulow, H. E. & Hobert, O. (2004). “Differential Sulfations and Epimerization Define Heparan Sulfate Specificity in Nervous System Development,” Neuron, 41, (5) 723–736.
Publisher – Google Scholar
Ciafre, S. A., Galardi, S., Mangiola, A., et al. (2005). “Extensive Modulation of a Set of Micrornas in Primary Glioblastoma,” Biochemical and Biophysical Research Communications, 334, (4) 1351–1358.
Publisher – Google Scholar
Conti, A., Aguennouz, M., La Torre, D., et al. (2009). “Mir-21 and 221 Upregulation and Mir-181b Downregulation in Human Grade II-IV Astrocytic Tumors,” Journal of Neuro-Oncology, 93, (3) 325–332.
Publisher – Google Scholar
Corsten, M. F., Miranda, R., Kasmieh, R., Krichevsky, A. M., Weissleder, R. & Shah, K. (2007). “Microrna-21 Knockdown Disrupts Glioma Growth in Vivo and Displays Synergistic Cytotoxicity with Neural Precursor Cell-Delivered S-TRAIL in Human Gliomas,” Cancer Research, 67, (19), 8994–9000.
Publisher – Google Scholar
Dechantsreiter, M. A., Planker, E., Matha, B., Lohof, E., Ho¨Lzemann, G., Jonczyk, A., et al. (1999). “N-Methylated Cyclic RGD Peptides as Highly Active and Selective Avb3 Integrin Antagonists,” American Chemical Society, 42, 3033–3040.
Publisher – Google Scholar
Derksen, P. W. B., Keehnen, R. M. J., Evers, L. M., Van Oers, M. H. J., Spaargaren, M. & Pals, S. T. (2002). “Cell Surface Proteoglycan Syndecan-1 Mediates Hepatocyte Growth Factor Binding and Promotes Met Signaling in Multiple Myeloma,” Blood, 99, (4) 1405–1410.
Publisher – Google Scholar
Dhoot, G. K., Gustafsson, M. K., Ai, X., Sun, W., Standiford, D. M. & Emerson, C. P. (2001). “Regulation of Wnt Signaling and Embryo Patterning by an Extracellular Sulfatase,” Science, 293, (5535) 1663-1666.
Publisher – Google Scholar
Du, J., Bernasconi, P., Clauser, K. R., et al. (2009). “Bead-Based Profiling of Tyrosine Kinase Phosphorylation Identifies SRC as a Potential Target for Glioblastoma Therapy,” Nature Biotechnology, (27) 77-83.
Publisher – Google Scholar
Egeblad, M. & Werb, Z. (2002). “New Functions for the Matrix Metalloproteinases in Cancer Progression,” Nature Reviews Cancer, 2, 161-174.
Publisher – Google Scholar
Eilers, M., Roy, U., Mondal, D. (2008). “MRP (ABCC) Transporters-Mediated Efflux Of Anti-HIV Drugs, Saquinavir And Zidovudine, From Human Endothelial Cells,” Experimental Biology and Medicine, 233, 1149-1160.
Publisher – Google Scholar
Esko, J. D. & Lindahl, U. (2001). “Molecular Diversity of Heparan Sulfate,” Journal of Clinical Investigation, 108, (2) 169–173.
Publisher – Google Scholar
Faria, C. C., Smith, C. A. & Rutka, J. T. (2013). “New Molecular Targets and Treatments for Pediatric Brain Tumors,” In Tech, 20, 555-573.
Publisher – Google Scholar
Feyzi, E., Lustig, F., Fager, G., Spillmann, D., Lindahl, U. & Salmivirta, M. (1997). “Characterization of Heparin and Heparan Sulfate Domains Binding to the Long Splice Variant of Platelet-Derived Growth Factor a Chain,” Journal of Biological Chemistry, 272, (9) 5518-5524.
Publisher – Google Scholar
Fillmore, H. L., Vanmeter, T. E. & Broaddus, W. C. (2001). “Membrane-Type Matrix Metalloproteinases (MT-Mmps): Expression and Function during Glioma Invasion,” Journal of Neuro-Oncology, 53, 187-202.
Publisher – Google Scholar
Fletcher, J. I., Haber, M., Henderson, M. J. & Norris, M. D. (2010). “ABC Transporters in Cancer: More Than Just Drug Efflux Pumps,” Nature Reviews Cancer, 10, 147–156.
Publisher – Google Scholar
Gallagher, J. T. (2001). “Heparan Sulfate: Growth Control with a Restricted Sequence Menu,” Journal of Clinical Investigation, 108, (3) 357- 361.
Publisher – Google Scholar
Gerstner, E. R., Yip, S., Wang, D. L., Louis, D. N., Iafrate, A. J. & Batchelor, T. T. (2009). “MGMT Methylation is a Prognostic Biomarker in Elderly Patients with Newly Diagnosed Glioblastoma,” Neurology, 73, 1509-1510.
Publisher – Google Scholar
Habuchi, H., Habuchi, O. & Kimata, K. (2004). “Sulfation Pattern in Glycosaminoglycan: Does it Have a Code?,” Glycoconjugate Journal, 21, (1-2) 47–52.
Publisher – Google Scholar
Hagerstrand, D., Hesselager, G., Achterberg, S., et al. (2006). “Characterization of an Imatinib-Sensitive Subset of High-Grade Human Glioma Cultures,” Oncogene, 25, 4913-4922.
Publisher – Google Scholar
Hartz, A. M. S & Bauer, B. (2010). “Regulation of ABC Transporters at the Blood-Brain Barrier: New Targets for CNS Therapy,” Molecular Interventions, 10, (5) 293-304.
Publisher – Google Scholar
Hayashida, K., Parks, W. C. & Park, P. W. (2009). “Syndecan-1 Shedding Facilitates the Resolution of Neurotrophic Inflammation by Removing Sequestered CXC Chemokines,” Blood, 114, (14) 3033-3043.
Publisher – Google Scholar
Hegi, M. E., Diserens, A. C., Gorlia, T., Hamou, M. F., Tribolet, N., Weller, M., et al. (2005). “MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma,” The New England journal of medicine, 352, 997–1003.
Publisher – Google Scholar
Heimberger, A. B., Hlatky, R., Suki, D., Yang, D., Weinberg, J., Gilbert, M., et al. (2005). “Prognostic Effect of Epidermal Growth Factor Receptor and Egfrviii in Glioblastoma Multiforme Patients,” Clinical Cancer Research, 11, 1462–1466.
Publisher – Google Scholar
Hong, X., Nelson, K. K., Decarvalho, A. C. & Kalkanis, S. N. (2010). “Heparanase Expression of Glioma in Human and Animal Models,” Journal of Neurosurgery, 113, (2) 261–269.
Publisher – Google Scholar
Hormigo, A., Ding, B.- S. & Rafiia, S. (2011). “A Target for Antiangiogenic Therapy: Vascular Endothelium Derived from Glioblastoma,” Proceedings of the National Academy of Sciences of the United States of America, 108, (11) 4271–4272.
Publisher – Google Scholar
Hur, J. H., Park, M. J., Park, I. C., Yi, D. H., Rhee, C. H., Hong, S. I., et al. (2000). “Matrix Metalloproteinases in Human Gliomas: Activation of Matrix Metalloproteinase-2 (MMP-2) May be correlated with Membrane-Type-1 Matrix Metalloproteinase (MT1-MMP) Expression,” Journal of Korean Medical Science, 15, 309-314.
Publisher – Google Scholar
Jansen, G., Scheper, R. J. & Dijkmans, B. A. (2003). “Multidrug Resistance Proteins in Rheumatoid Arthritis, Role in Disease-Modifying Antirheumatic Drug Efficacy and Inflammatory Processes: An Overview,” Scandinavian Journal of Rheumatology, 32, 325-336.
Publisher – Google Scholar
Kleihues, P. & Ohgaki, H. (1999). “Primary and Secondary Glioblastomas: From Concept to Clinical Diagnosis,” Neuro-Oncology, 1, 44–51.
Publisher – Google Scholar
Kreuger, J., Salmivirta, M., Sturiale, L., Gimenez-Gallego, G. & Lindahl, U. (2001). “Sequence Analysis of Heparan Sulfate Epitopes with Graded Affinities for Fibroblast Growth Factors 1 and 2,” The Journal of Biological Chemistry, 276, (33) 30744-30752.
Publisher – Google Scholar
Kuan, C. T., Wakiya, K., Herndon, J. E., Lipp, E. S., Pegram, C. N. & Riggins, G. J. (2010). “MRP3; A Molecular Target for Human Glioblastoma Multiforme Immunotherapy,” BMC Cancer, 10, (468) 1-15.
Publisher – Google Scholar
Kulasingam, V. & Diamandis, E. (2008). “Strategies for Discovering Novel Cancer Biomarkers through Utilization of Emerging Technologies,” Nature Clinical Practice Oncology, 5, (10) 588-599.
Publisher – Google Scholar
Kurokawa, H., Katsube, K., Podyma, K. A., Ikuta, M., Iseki, H., Nakajima, M., Akashi, T., Omura, K., Takagi, M. & Yanagishita, M. (2003). “Heparanase and Tumor Invasion Patterns in Human Oral Squamous Cell Carcinoma Xenografts,” Cancer Science, 94, (3) 277–285.
Publisher – Google Scholar
Lages, E., Guttin, A., Atifi, M. E., et al. (2011). “Microrna and Target Protein Patterns Reveal Physiopathological Features of Glioma Subtypes,” PLoS One, 6, (5) E20600.
Publisher – Google Scholar
Lampert, K., Machein, U., Machein, M. R., Conca, W., Peter, H. H. & Volk, B. (1998). “Expression of Matrix Metalloproteinases and Their Tissue Inhibitors in Human Brain Tumors,” The American Journal of Pathology, 153, 429-437.
Publisher – Google Scholar
Liang, Y., Diehn, M., Watson, N., Bollen, A. W., Aldape, K. D., Nicholas, M. K., et al. (2005). “Gene Expression Profiling Reveals Molecularly and Clinically Distinct Subtypes of Glioblastoma Multiforme,” Proceedings of the National Academy of Sciences of the United States of America, 102, 5814–5819.
Publisher – Google Scholar
Liu, C., Sage, J. C., Miller, M. R., Verhaak, R. G. W., Hippenmeyer, S., Vogel, H., et al. (2011). “Mosaic Analysis with Double Markers Reveals Tumor Cell of Origin in Glioma,” Cell, 146, 1–13.
Publisher – Google Scholar
Llano, E., Pendas, A. M., Freije, J. P., Nakano, A., Knauper, V., Murphy, G., et al. (1999). “Identification and Characterization of Human MT5-MMP, a New Membrane-Bound Activator of Progelatinase a Overexpressed in Brain Tumors,” Cancer Research, 59, 2570-2576.
Publisher – Google Scholar
Louis, D. N., Ohgaki, H., Wiestler, O. D. et al. (2007). “The 2007 WHO Classification of Tumours of the Central Nervous System,” Acta Neuropathologica, 114, (2) 97-109.
Publisher – Google Scholar
Macdonald, T. J., Stewart, C. F., Kocak, M., Goldman, S., Ellenbogen, R. G., Phillips, P., et al. (2008). “Phase I Clinical Trial of Cilengitide in Children with Refractory Brain Tumors: Pediatric Brain Tumor Consortium Study PBTC- 012,” Journal of Clinical Oncology, 26, (6) 919–924.
Publisher – Google Scholar
Macdonald, T. J., Taga, T., Shimada, H., Tabrizi, P., Zlokovic, B. V., Cheresh, D. A., et al. (2001). “Preferential Susceptibility of Brain Tumors to the Antiangiogenic Effects of an Alpha (V) Integrin Antagonist,” Neurosurgery, 48, 151–157.
Publisher – Google Scholar
Mclendon, R., Friedman, A., Bigner, D. & Vanmeir, E. (2008). “Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways,” Nature, 455, 1061-1068.
Publisher – Google Scholar
Mikkelsen, T., Brodie, C., Finniss, S., Berens, M. E., Rennert, J. L., Nelson, K., et al. (2008). “Radiation Sensitization of Glioblastoma by Cilengitide Has Unanticipated Scheduledependency,” Journal of Cancer, 124, 2719–27.
Publisher – Google Scholar
Miller, D. S., Bauer, B. & Hartz, A. M. S. (2008). “Modulation of P-Glycoprotein at the Blood-Brain Barrier: Opportunities to Improve Central Nervous System Pharmacotherapy,” Pharmacological Reviews, 60, 196–209.
Publisher – Google Scholar
Mischel, P. S. & Cloughesy, T. F. (2003). “Targeted Molecular Therapy for GBM,” Brain Pathology, 13, 52-61.
Publisher – Google Scholar
Nabors, L. B., Mikkelsen, T., Rosenfeld, S. S., Hochberg, F., Akella, N. S., Fisher, J. D., et al. (2007). “Phase I and Correlative Biology Study of Cilengitide in Patients with Recurrent Malignant Glioma,” Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 25, (13) 1651–1657.
Publisher – Google Scholar
Nagase, H., Visse, R. & Murphy, G. (2006). “Structure and Function of Matrix Metalloproteinases and Timps,” Cardiovascular Research, 69, 562-573.
Publisher – Google Scholar
Nakada, M., Nakamura, H., Ikeda, E., Fujimoto, N., Yamashita, J., Sato, H., et al. (1999). “Expression and Tissue Localization of Membrane-Type 1, 2, and 3 Matrix Metalloproteinases in Human Astrocytic Tumors,” The American Journal of Pathology, 154, 417-428.
Publisher – Google Scholar
Nakagawa, T., Kubota, T., Kabuto, M., Sato, K., Kawano, H., Hayakawa, T., et al. (1994). “Production of Matrix Metalloproteinases and Tissue Inhibitor of Metalloproteinases-1 by Human Brain Tumors,” Journal of Neurosurgery, 81, 69-77.
Publisher – Google Scholar
Nakano, A., Tani, E., Miyazaki, K., Yamamoto, Y. & Furuyama, J. (1995). “Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Human Gliomas,” Journal of Neurosurgery, 83, 298-307.
Publisher – Google Scholar
Ono, K., Hattori, H., Takeshita, S., Kurita, A. & Ishihara, M. (1999). “Structural Features in Heparin That Interact with VEGF165 and Modulate Its Biological Activity,” Glycobiology, 9, (7) 705-711.
Publisher – Google Scholar
Perry, J., Okamoto, M., Guiou, M., Shirai, K., Errett, A. & Chakravarti, A. (2012). “Novel Therapies in Glioblastoma,”Neurology Research International, 1-14.
Publisher – Google Scholar
Phillips, H. S., Kharbanda, S., Chen, R., Forrest, W. F., Soriano, R. H., Wu, T. D., et al. (2006). “Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis,” Cancer Cell, 9, 157–173.
Publisher – Google Scholar
Phillips, H. S., Kharbanda, S., Chen, R., Forrest, W. F., Soriano, R. H., Wu, T. D., Misra, A., Nigro, J. M., Colman, H., Soroceanu, L., Williams, P. M., Modrusan, Z., Feuerstein, B. G. & Aldape K. (2006). “Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis,” Cancer Cell, 9, (3) 157-173.
Publisher – Google Scholar
Phillips, J. J. (2012). “Novel Therapeutic Targets in the Brain Tumor Microenvironment,” Oncotarget, 3, (5) 568-575.
Publisher – Google Scholar
Phillips, J. J., Huillard, E., Robinson, A. E., Ward, A., Lum, D. H., Polley, M. Y., Rosen, S. D., Rowitch, D. H. & Werb, Z. (2012). “Heparan Sulfate Sulfatase SULF2 Regulates PDGFR-Alpha Signaling and Growth in Human and Mouse Malignant Glioma,” The Journal of Clinical Investigation, 122, (3) 911–922.
Publisher – Google Scholar
Ramani, V. C., Yang, Y., Ren, Y., Nan, L. & Sanderson, R. D. (2011). “Heparanase Plays a Dual Role in Driving Hepatocyte Growth Factor (HGF) Signaling by Enhancing HGF Expression and Activity,” The Journal of Biological Chemistry, 286, (8) 6490–6499.
Publisher – Google Scholar
Rapraeger, A. C. (2000). “Syndecan-Regulated Receptor Signaling,” The Journal of Cell Biology, 149, (5) 995-998.
Publisher – Google Scholar
Ridgway, L. D., Wetzel, M. D. & Marchetti, D. (2011). “Heparanase Modulates Shh and Wnt3a Signaling in Human Medulloblastoma Cells,” Experimental and Therapeutic Medicine, 2, (2) 229–238.
Publisher – Google Scholar
Rieger, J., Lemke, D., Maurer, G., et al. (2008). “Enzastaurin-Induced Apoptosis in Glioma Cells is Caspase-Dependent and Inhibited by BCL-XL,” Journal of Neurochemistry, (106) 2436-2448.
Publisher – Google Scholar
Ruano, Y., Mollejo, M., Ribalta, T., Fiano, C., Camacho, F. I., Gomez, E., et al. (2006). “Identification of Novel Candidate Target Genes in Amplicons of Glioblastoma Multiforme Tumors Detected by Expression and CGH Microarray Profiling,” Molecular Cancer, 5, 39.
Publisher – Google Scholar
Sarkadi, B., Homolya, L., Szakacs, G. & Varadi, A. (2006). “Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System,” Physiological Reviews, 86, 1179-1236.
Publisher – Google Scholar
Sarrazin, S., Lamanna, W. C. & Esko, J. D. (2011). “Heparan Sulfate Proteoglycans,” Cold Spring Harbor Perspectives in Biology, 3(7).
Publisher – Google Scholar
Sasisekharan, R., Shriver, Z., Venkataraman, G. & Narayanasami, U. (2002). “Review Roles of Heparan-Sulphate Glycosaminoglycans in Cancer,” Nature Reviews Cancer, 2, (7) 521-528.
Publisher – Google Scholar
Seidal, C., Sundan, A., Hjorth, M., Turesson, I., Dahl, I. M., Abildgaard, N., et al. (2000). “Serum Syndecan-1: A New Independent Prognostic Marker in Multiple Myeloma,” Blood, 95, (2) 388-392.
Publisher – Google Scholar
Shai, R., Shi, T., Kremen, T. J., Horvath, S., Liau, L. M., Cloughesy, T. F., et al. (2003). “Gene Expression Profiling Identifies Molecular Subtypes of Gliomas,” Oncogene, 22, 4918–4923.
Publisher – Google Scholar
Shen, S. & Zhang, W. (2010). “ABC Transporters and Drug Efflux at the Blood-Brain Barrier,” Reviews in the Neurosciences, 21, 29–53.
Publisher – Google Scholar
Shinojima, N., Tada, K., Shiraishi, S., Kamiryo, T., Kochi, M., Nakamura, H., et al. (2003). “Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme,” Cancer Research, 63, 6962–6970.
Publisher – Google Scholar
Slaby, O., Lakomy, R., Fadrus, P., et al. (2010). “Microrna-181 Family Predicts Response to Concomitant Chemoradiotherapy with Temozolomide in Glioblastoma Patients,” Neoplasma, 57, (3) 264–269.
Publisher – Google Scholar
Smith, E. M., Mitsi, M., Nugent, M. A. & Symes, K. (2009). “PDGF-A Interactions with Fibronectin Reveal a Critical Role for Heparan Sulfate in Directed Cell Migration during Xenopus Gastrulation,” Proceedings of the National Academy of Sciences of the United States of America, 106, (51) 21683-21688.
Publisher – Google Scholar
Spence, A. M., Muzi, M., Graham, M. M., et al. (1998). “Glucose Metabolism in Human Malignant Gliomas Measured Quantitatively with PET, 1-[C-11] Glucose and FDG: Analysis of the FDG Lumped Constant,” Journal of Nuclear Medicine : Official Publication, Society of Nuclear Medicine, 39, 440-448.
Publisher – Google Scholar
Stupp, R., Mason, W. P., Van Den Bent, M. J., et al. (2005). “Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma,” The New England Journal of Medicine, 352, 987-996.
Publisher – Google Scholar
The Cancer Genome Atlas Research Network. (2008). 455, (23) 1061-1068.
Toschi, L. & Janne, P. A. (2008). “Single-Agent and Combination Therapeutic Strategies to Inhibit Hepatocyte Growth Factor/MET Signaling in Cancer,” Clinical Cancer Research, 14, 5941-5946.
Publisher – Google Scholar
Towner, R. A., He, T., Doblas, S. & Smith, N. (2011). “Assessment of Rodent Glioma Models Using Magnetic Resonance Imaging Techniques,” In Tech, 251-272.
Publisher – Google Scholar
Tso, C. L., Freije, W. A., Day, A., Chen, Z., Merriman, B., Perlina, A., et al. (2006). “Distinct Transcription Profiles of Primary and Secondary Glioblastoma Subgroups,” Cancer Research, 66, 159–167.
Publisher – Google Scholar
Tusher, V. G., Tibshirani, R. & Chu, G. (2001). “Significance Analysis of Microarrays Applied to the Ionizing Radiation Response,” Proceedings of the National Academy of Sciences of the United States of America, 98, 5116–5121.
Publisher – Google Scholar
Ujifuku, K., Mitsutake, N., Takakura, S., et al. (2010). “miR-195, miR-455-3p and miR-10a* are Implicated in Acquired Temozolomide Resistance in Glioblastoma Multiforme Cells,” Cancer Letters, 296, (2) 241–248.
Publisher – Google Scholar
Velasco, G., Cal, S., Merlos-Suarez, A., Ferrando, A. A., Alvarez, S., Nakano, A., et al. (2000). “Human MT6-Matrix Metalloproteinase: Identification, Progelatinase a Activation, and Expression in Brain Tumors,” Cancer Research, 60, 877-882.
Publisher – Google Scholar
Wachsberger, P. R., Lawrence, Y. R., Liu, Y., Daroczi, B., Xu, X. & Dicker, A. P. (2012). “Epidermal Growth Factor Receptor Expression Modulates Antitumor Efficacy of Vandetanib or Cediranib Combined with Radiotherapy in Human Glioblastoma Xenografts,” International Journal of Radiation Oncology*Biology*Physics, 82, (1) 483–491.
Publisher – Google Scholar
Wade, A., Robinson, A. E., Engler, J. R., Petritsch, C., James, C. D. & Phillips, J. J. (2013). “Proteoglycans and Their Roles in Brain Cancer,” FEBS Journal, 280, (10) 2399-2417.
Publisher – Google Scholar
Whiteford, J. R., Xian, X., Chaussade, C., Vanhaesebroeck, B., Nourshargh, S. & Couchman, J. R. (2011). “Syndecan-2 is a Novel Ligand for the Protein Tyrosine Phosphatase Receptor CD148,” Molecular Biology of the Cell, 22, (19) 3609-3624.
Publisher – Google Scholar
Wikstrand, C. J., Bigner, S. H. & Bigner, D. D. (1983). “Demonstration of Complex Antigenic Heterogeneity in a Human Glioma Cell Line and Eight Derived Clones by Specific Monoclonal Antibodies,” Cancer Research, 43, 3327-3334.
Publisher – Google Scholar
Xiong, J. P., Stehle, T., Zhang, R., Joachimiak, A., Frech, M., Goodman, S. L., et al. (2002). “Crystal Structure of the Extracellular Segment of Integrin Alphav Beta3 in Complex with an Arg-Gly-Asp Ligand,” Science, 296, 151–155.
Publisher – Google Scholar
Yang, Y., Yaccoby, S., Liu, W., Langford, J. K., Pumphrey, C. Y., Theus, A., et al. (2002). “Soluble Syndecan-1 Promotes Growth of Myeloma Tumors in Vivo,” Blood, 100, (2) 610-617.
Publisher – Google Scholar