Introduction
Schistosomiasis is a common parasitic disease that ranks second only to malaria as a global cause of morbidity and mortality. It occurs in 76 tropical and subtropical countries and affects about 10% of the world’s population. The prevalence of this infection remains high despite the availability of efficacious antischistosomal drugs. Individuals become infected with the parasite at an early age by exposure to the free-swimming larval stage (termed cercariae) in contaminated water (Rosin et al, 1994). Schistosomiasis is the first in prevalence among all water-borne infectious diseases and one of the main occupational risks encountered in the rural endemic areas of the world. Schistosomiasis-associated bladder cancer defines a characteristic pathological and histogenic nature that differentiates it from bladder cancer of non-schistosomal origin. It shows an increased incidence of squamous cell carcinoma as opposed to transitional cell carcinoma, a lower mean age at diagnosis, and a higher male/female ratio. Moreover, in association with schistosomiasis, cancer may originate throughout the bladder but rarely in the trigone, a frequent site of tumor origin in non-schistosomal cases (Badawi, 1996). Human bladder cancer is the fifth to the seventh most common cancer in Western countries. In many tropical and subtropical areas, however, it is the first among all types of cancer, mainly due to the endemic parasitism (Badawi, 1996).
Many oncogenes participate in the development of human tumors. The most frequently activated genes are members of the RAS gene family, two of which (Harvey- H) and (Kirsten-K) were first identified as being homologous to viral transforming genes, while the third (N-ras) was identified in a human neuroblastoma. The RAS gene product is a monomeric membrane-localized G protein of 21 Kd that functions as a molecular switch linking receptor and nonreceptor tyrosine kinase activation to downstream cytoplasmic or nuclear events. Each mammalian cell contains at least 3 distinct RAS proto-oncogenes encoding closely related, but distinct proteins. RAS genes can be activated by point mutation at codons 12, 13, 61, or 113-117. Signal transduction by Ras proteins involves hydrolysis of GTP, and activating mutations inhibit this process, locking the protein in the “on” signaling conformation (Stanley, 1995).
Activating mutations in these Ras proteins result in constitutive signaling, thereby stimulating cell proliferation and inhibiting apoptosis. Oncogenic mutations in the RAS genes are present in approximately 30% of all human cancers (Adjei, 2001). The high degree of similarities shared by different Ras isoforms suggests that functional redundancy exists among these proteins, yet accumulating data support for unique roles of the Ras family members with K-ras being the most important Ras isoform (Zhang et al, 2007). Oncogenic KRAS mutations are prevalent in virtually all cancer types, making KRAS one of the most frequently mutated genes in human cancers (Zhang et al, 2007).
Mutational activation of RAS protooncogenes is frequently observed in human tumors and in experimental animals (Cerutti P et al, 1994). KRAS mutations have been found in bladder cancer patients (Jebar et al, 2005; Gormally et al, 2006), in transformed bladder epithelial cells (Brookes et al, 1988) and chemically-induced rat bladder tumors (Stanley, 1995).
We have previously demonstrated that normal CHO (Chinese Hamster Ovary) cells treated in laboratory with S. haematobium total antigen, acquire cancer-like characteristics. In fact, these cells presented rapid uncontrolled division, high resistance to death and an abnormal capability to migrate (Botelho et al, 2009a) and, when injected into nude mice, lead to tumour development. Crucially, animals injected with non-exposed cells showed no growth (Botelho et al, 2009b). We have further used CD-1 mice to show that Schistosoma haematobium total antigen (Sh) has a carcinogenic ability. Sh intravesically instillation induced the development of dysplasia in 70% of animals sacrificed 40 weeks after. Our results suggest that Sh induce the malignization of the urothelium (Botelho et al, 2011). Dysplasia (LGIUN— Low Grade Intra-Urothelial Neoplasia) is preneoplastic lesion that can evolutes to carcinoma in situ, invasive carcinoma and/or papillary carcinoma. In the present study, we addressed gene mutations that constitutively activate the receptor tyrosine kinase-Ras pathway and therefore we studied activating mutations in KRAS gene. Twenty percent of the bladders with dysplasia presented a KRAS mutation in codon 12 of exon 2. We concluded from these results that the parasite extract of S. haematobium may have carcinogenic ability possibly through oncogenic mutation of KRAS gene.
An improved understanding of the molecular basis underlying bladder tumor pathways and specifically schistosomiasis-associated bladder cancer is of clear importance in helping to better predict bladder tumor progression and devise optimal management strategies for this type of carcinoma.
Material and Methods
Animals
Eight-week-old female golden hamsters (LVG/SYR) and CD-1 mice were provided by Charles River (Barcelona, Spain). Animals spent one week being acclimated under routine laboratory conditions before starting the experiments. They did not receive any treatment prior to the study. Hamsters were kept in separated cages and mice were kept in 6 litter mates cage. They were fed standard balanced food and water ad libitum. All the animals were maintained at the National Institute of Health (Porto, Portugal) in rooms with controlled temperature (22 ± 2ºC) and humidity (55% ± 10%) and continuous air renovation. Animals were housed in a 12 h light/12 h dark cycle (8 am-8 pm). All animal experiments were performed in accordance with the National (DL 129/92; DL 197/96; P 1131/97) and European Convention for the Protection of Animals used for Experimental and Other Scientific Purposes and related European Legislation (OJ L 222, 24.8.1999).
Experimental Infections
Urine was collected from S. haematobium-infected individuals. The individuals were living in Angola, an endemic area for schistosomiasis. Informed consent from patients was obtained. S. haematobium infection was detected by microscopically observation of the eggs in the sediment of centrifuged urine. The eggs were hatched and with the resulting miracidia, snails from susceptible species were infected.
Golden hamsters were experimentally infected with 100 cercariae by tail immersion. The control animals consisted of littermates. The cercariae were obtained by shedding of snails infected with miracidia.
S. Haematobium Total Antigen (Sh) Production
Angolan strain of S. haematobium adult worms were collected by perfusion of the hepatic portal system of golden hamsters at seven weeks after infection with 100 cercarie. The worms were suspended in PBS and then sonicated. The protein extract was then ultracentrifuged and the protein concentration was estimated using a micro BCA protein assay reagent kit.
Experimental Design
Forty mice were randomly divided into six experimental groups. Groups 1 and 3 (n=10) were Sh-instilled animals. Groups 2 (n=5) and 4 (n=5) were control animals with their bladders instilled with saline only. Groups 1 and 2 were sacrificed 20 weeks after instillation; group 3 and 4 were sacrificed 40 weeks after instillation. For the duration of the study, the mice’s state of health was monitored daily.
For intravesical instillation of the parasite total antigen, 50 g/ml of Sh was introduced directly into the urinary bladders of female mice via the urethra according to the procedure previously described (Chin et al, 1996). Briefly, animals were anesthetized with an intraperitoneal injection of sodium pentobarbital (40 mg/kg). The bladder was catheterized via the urethra with a 24 gauge plastic intravenous cannula and the bladder mucosa was then traumatized to remove the umbrella cells of the urothelium by instilling 0.1 ml of 0.1N HCl solution for 15 seconds, neutralized with 0.1 ml of 0.1N KOH and flushed with sterile saline.
A total of 150 l of Sh with a concentration of 50 g/ml was instilled via the cannula. The dwell time after instillation was approximately 1 hour. The mice were turned 90 degrees every 15 minutes to facilitate whole bladder exposure to the Sh intravesically instilled. One hour after the instillation, micturition of the mice was induced by light abdominal massage so that the duration of exposure was constant. Body temperature was maintained with a homoeothermic bandage. All the experiments described herein were carried out under aseptic conditions and performed once during this study.
All animals were sacrificed by intraperitoneal administration of sodium pentobarbital (130 mg/Kg). Complete necropsies were carefully conducted. All organs were examined macroscopically for any changes. The urinary bladders were inflated in situ by injection of 10% phosphate buffered formalin (150μl), ligated around the neck to maintain proper distension and then were immersed in the same solution for 12 hours. After fixation, the formalin was removed; the urinary bladder was cut into two strips, and routinely processed. Tissue sections (2 μm) from urinary bladder, spleen, liver and lung were stained with haematoxylin and eosin (HE) to characterize changes and inflammatory infiltrate.
Mutation Analysis of KRAS Gene
Whole tissue of the bladders was used for mutational analysis of KRAS codon 12. Direct sequencing was performed and Big Dye Terminator (version 1.1) was used according to instructions of supplier (Applied Biosystems).
Results
Animals’ General Aspect
During the experimental work no animal died and all, animals exhibited normal cage activity. The weight of the animals remained normal throughout the experiment (data not shown).
Macroscopic and Microscopic Evaluation
The bladders from all groups showed no gross lesions. No macroscopic changes were seen in the liver, lungs or kidneys of all animals. The mean liver, kidney and bladder weights, as well as relative liver, kidney and bladder weights at the end of the study did not show differences (data not shown).
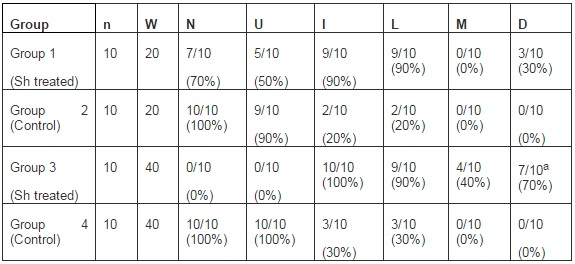
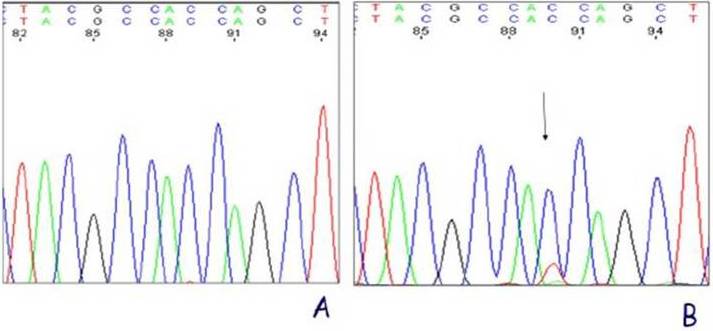
The bladder histopathological findings observed in all groups are given in Table 1 and Figure 1. Bladder histopathology of animals instilled with Sh was characterized by extensive diffuse or nodular infiltrate composed predominantly of lymphocytes admixed with variable number of eosinophils, and by absence of umbrella cells, nodular hyperplasia and dysplasia. The nodular hyperplasia exhibiting budlike proliferated urothelial cells extending into the lamina propria. After 20 weeks of treatment 30% of these animals displayed urothelial dysplasia a LGIUN (Low Grade Intra Urothelial Neoplasia), whereas after 40 weeks of treatment we found 70% of the bladders with this LGIUN, a non-invasive malignant flat lesion in the urothelium of these animals. Mastocytes were only found in the bladders of animals sacrificed after 40 weeks. No bladder histopathological changes were observed in animals from control groups. No microscopic changes were seen in the spleen, lung, kidney and liver in samples obtained from all animals.
Table 1: Histopathological Findings in the Sh Instilled Bladders and Controls
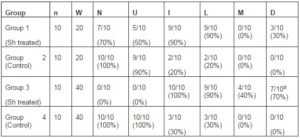
n, number of animals; W, weeks between treatment and sacrifice; N, normal urothelium; U, presence of umbrella cells; I, inflammatory infiltrate; L, infiltration of lymphocyte; M, infiltration of mastocytes; D, dysplasia. a (p=0.001, Group 3 Vs. Group 4) (12).
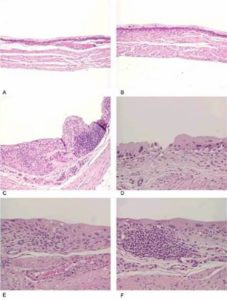
Figure 1: (A and B) Control, (C) Urothelium Displaying an Inflammatory Nodule and a Lesion with Characteristics of Nodular Hyperplasia, (D) Denudation and Dysplasia, (E) Inflammatory Infiltrate and Dysplasia, (F) Inflammatory Nodule and Dysplasia (12)
Frequency of KRAS Mutations in Codon 12
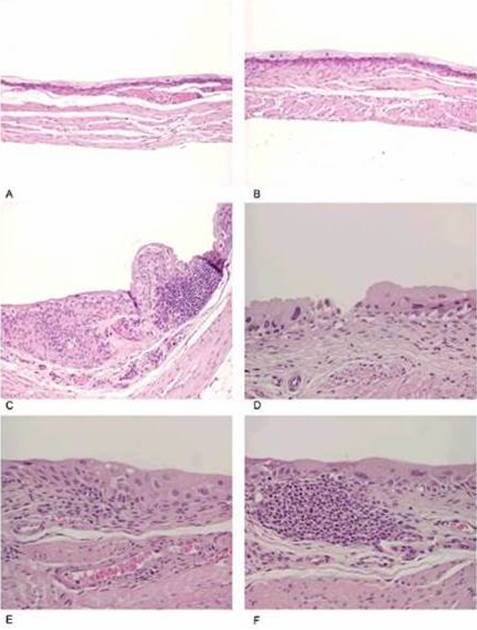
Two (out of 10) of the bladders with dysplasia presented a KRAS mutation in codon 12 of exon 2 (Fig. 2). This results in Gly12Asp mutation. The controls showed no mutation. We concluded from these results that the parasite extract of S. haematobium has carcinogenic ability possibly through oncogenic mutation of KRAS gene.
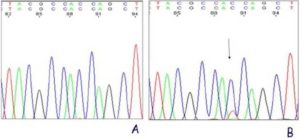
Figure 2: KRAS Exon 2 Sequencing Results. (A) Control, (B) Dysplastic Bladder with a Mutation at Codon 12 (Resulting in a Gly12Asp Mutation).
Discussion
Bladder cancer is a complex disease, with a multitude of genetic aberrations already uncovered, and likely more still to come. We previously demonstrated that Schistosoma haematobium total antigen has a carcinogenic ability in a CD-1 mice model. We showed that intravesically administration of Sh gave rise to a high incidence of urothelial dysplasia, a non-invasive malignant flat lesion, in 70% of the tested subjects after 40 weeks of treatment. In the present study, we were able to investigate this effect further and show that 20% of the dysplastic bladders presented a mutation in codon 12 of KRAS gene.
It is generally recognized that the pathogenesis of malignancy involves three distinct and sequential effects on the target cells; initiation, promotion and progression. The first stage is thought to involve the formation of damage in the DNA resulting in a somatic cell mutation. The essential feature of promotion, however, is the induction of proliferation in the initiated cells by agents that are not necessarily carcinogenic per se (Gutiérrez and Salsamendi, 2001). The sequence of lesions identified, via histopathology, between initiation and promotion are designated as preneoplastic lesions and/or benign neoplasias (Gutiérrez and Salsamendi, 2001). Their transformation into malignant lesions is the last of the stages of carcinogenesis and is the most extended – it is labelled progression (Klaunig et al, 2000; Williams, 2001). We cannot state at this point that KRAS mutation that we detected in dysplastic bladders induced by Sh treatment is an initiating event. The presence of this mutation could have one of two explanations: either the parasite total antigen has carcinogenic molecule(s) sufficiently genotoxic and, consistent with the mutation observed, caused direct attack on the RAS gene leading to uncontrolled proliferation of the cells and causing dysplasia in the bladders treated with the parasite antigen; or the parasite antigens are only mitogenic for the urothelial cells causing increased proliferation that resulted in the KRAS mutation via non-carcinogen-mediated pathways.
In agreement with the first explanation other researchers have well characterized the phenomenon of RAS oncogene activation in a number of systems. In some cases, such as the mouse and rat mammary gland, RAS activation is clearly an important initiating event, and when tumors are induced by genotoxic carcinogens the mutations observed are consistent with direct attack by the carcinogen on the RAS gene (Stanley, 1995). Also the majority of evidence taken from several animal studies suggests that RAS proto-oncogene activation is an early event. This presumption is derived from detection of RAS activation in benign (early) tumors of skin, lung and liver. Based upon tumor induction following a brief exposure to carcinogens that produce a chemical-specific point mutation in RAS activation, researchers have suggested that activation of RAS may be the initiating event in some model systems (Moronpot et al, 1995). Furthermore, the activated RAS gene has been detected in many benign tumors, including mouse skin papillomas, mouse lung and liver adenomas, and basal cell and clitoral gland tumors of the rat. This implies that the activated RASwas present in the cell that clonally expanded to these benign tumors. Thus, activation of the RAS proto-oncogene may be the initiation event in some model systems (Stowers et al, 1987).
Few studies were carried out to investigate the role and frequency of activated RAS in schistosomiasis-associated bladder cancer (Badawi, 1996). The mutations were not detected in two previous studies (Fugita et al, 1987; Ramchurren N et al, 1995). These findings indicate that the advanced stage of schistosomiasis-associated bladder cancer may not involve a high frequency of detectable mutations within the RAS genes. It is known that more than 90% of the SCC in the schistosome-infected bladder were advanced (stages T3 and T4) at the time of diagnosis. However, this does not exclude the possibility that activation in RAS could be an important factor in the initiation as well as promotion and progression of schistosomiasis-associated bladder cancer (Badawi, 1996).
Further studies are needed to address the functional effect of KRAS mutations and the clinical relevance of these mutations in schistosomiasis-associated bladder cancer.
Acknowledgements
We would like to express our deepest appreciation to Mrs. Lígia Lourenço for her technical assistance, Dr. Paulo Vieira and Mrs. Maria de Lurdes Delgado for animal care and antigen production.
(adsbygoogle = window.adsbygoogle || []).push({});
References
Adjei, A. A. (2001). “Blocking Oncogenic Ras Signaling for Cancer Therapy,” J Natl Cancer Inst 93: 1062-1074.
Publisher – Google Scholar – British Library Direct
Badawi, A. F. (1996). “Molecular and Genetic Events in Schistosomiasis-Associated Human Bladder Cancer: Role of Oncogenes and Tumor Suppressor Genes,” Cancer Lett 105: 123-138.
Publisher – Google Scholar
Botelho, M., Ferreira, A. C., Oliveira, M. J., Domingues, A., Machado, J. C. & da Costa, J. M. (2009). “Schistosoma Haematobium Total Antigen Induces Increased Proliferation, Migration and Invasion, and Decreases Apoptosis of Normal Epithelial Cells,” Int J Parasitol 39: 1083-1091.
Publisher – Google Scholar
Botelho, M., Oliveira, P., Gomes, J., Gartner, F., Lopes, C., Correia da Costa, J. M. & Machado, J. C. (2009). “Tumourigenic Effect of Schistosoma Haematobium Total Antigen in Mammalian Cells,” Int J Exp Path 90: 448-453.
Publisher – Google Scholar
Botelho, M. C., Oliveira, P. A., Lopes, C., Correia da Costa, J. M. & Machado, J. C. (2011). “Urothelial Dysplasia and Inflammation Induced by Schistosoma Haematobium Total Antigen Instillation in Mice Normal Urothelium,” Urol Oncol 29: 809-814.
Publisher – Google Scholar
Brookes, P., Cooper, C. S., Ellis, M. V., Warren, W., Gardner, E. & Summerhayes, I. C. (1988). “Activated Ki-Ras Genes in Bladder Epithelial Cell Lines Transformed by Treatment of Primary Mouse Bladder Explant Cultures with 7,12-Dimethylbenz[A]Anthracene,” Mol Carcinog. 1: 82-88.
Publisher – Google Scholar
Cerutti, P., Hussain, P., Pourzand, C. & Aguilar, F. (1994). “Mutagenesis of the H-Ras Protooncogene and the P53 Tumor Suppressor Gene,” Cancer Res 54: 1934s-1938s.
Publisher – Google Scholar – British Library Direct
Chin, J. L., Kadhim, S. A., Batislam, E., Karlik, S. J., Garcia, B. M., Nickel, J. C. & Morales, A. (1996). “Mycobacterium Cell Wall: An Alternative to Intravesical Bacillus Calmette Guerin (BCG) Therapy in Orthotopic Murine Bladder Cancer,” J Urol 156: 1189-1193.
Publisher – Google Scholar – British Library Direct
Fujita, J., Nakayama, H., Onoue, H., Rhim, J. S., El-Bolkainy, M. N., El-Aaser, A. A. & Kitamura, Y. (1987). “Frequency of Active Ras Oncogenes in Human Bladder Cancers Associated with Schistosomiasis,” Jpn J Cancer Res 78: 915-920.
Publisher – Google Scholar
Gormally, E., Vineis, P., Matullo, G., Veglia, F., Caboux, E., Le Roux, E., Peluso, M., Garte, S., Guarrera, S. &
Munnia, A. (2006). “TP53 and KRAS2 Mutations in Plasma DNA of Healthy Subjects and Subsequent Cancer Occurrence: A Prospective Study,” Cancer Res 66: 6871-6876.
Publisher – Google Scholar – British Library Direct
Gutiérrez, J. B. & de Cerain Salsamendi, A. L. (2001). Fundamentos de Ciência Toxicológica, Madrid: Diaz de Santos, Pp 155-177.
Publisher – Google Scholar
Jebar, A. H., Hurst, C. D., Tomlinson, D. C., Johnston, C., Taylor, C. F. & Knowles, M. A. (2005). “FGFR3 and Ras Gene Mutations are Mutually Exclusive Genetic Events in Urothelial Cell Carcinoma,” Oncogene 24: 5218-5225.
Publisher – Google Scholar – British Library Direct
Klaunig, J. E., Kamendulis, L. M. & Xu, Y. (2000). “Epigenetic Mechanisms of Chemical Carcinogenesis,” Hum Exp Toxicol 19: 543-555.
Publisher – Google Scholar – British Library Direct
Maronpot, R. R., Fox, T., Malarkey, D. E. & Goldsworthy, T. L. (1995). “Mutations in the Ras Proto-Oncogene: Clues to Etiology and Molecular Pathogenesis of Mouse Liver Tumors,” Toxicology 101: 125-156.
Publisher – Google Scholar
Ramchurren, N., Cooper, K. & Summerhayes, I. C. (1995). “Molecular Events Underlying Schistosomiasis-Related Bladder Cancer,” Int J Cancer 62: 237-244.
Publisher – Google Scholar – British Library Direct
Rosin, M. P., Anwar, W. A. & Ward, A. J. (1994). “Inflammation, Chromosomal Instability, and Cancer: The Schistosomiasis Model,” Cancer Res 54: 1929s-1933s.
Publisher – Google Scholar – British Library Direct
Stanley, L. A. (1995). “Molecular Aspects of Chemical Carcinogenesis: The Roles of Oncogenes and Tumour Suppressor Genes,” Toxicology 96: 173-194.
Publisher – Google Scholar
Stowers, S. J., Maronpot, R. R., Reynolds, S. H. & Anderson, M.W. (1987). “The Role of Oncogenes in Chemical Carcinogenesis,” Environ Health Perspect 75: 81.
Publisher – Google Scholar
Williams, G. M. (2001). “Mechanisms of Chemical Carcinogenesis and Application to Human Cancer Risk Assessment,” Toxicology 161: 3-10.
Publisher – Google Scholar
Zhang, J. & Lodish, H. F. (2007). “Endogenous K-Ras Signaling in Erythroid Differentiation,” Cell Cycle 6: 1970-1973.
Publisher – Google Scholar – British Library Direct