Discussion
The combined approach proposed here has proved its usefulness for allowing the rapid cloning and characterization of specific conserved genes, as well for providing plant genomic fingerprinting information. An orthologous element for the Arabidopsis More Axillary Branching gene was successfully cloned and sequenced, for the first time, from several H. rosa-sinensis cultivars and from Hibiscus wild species. This general approach is particularly useful when dealing with plant species for which no or poor information is available at the genomic level. Minor changes could be made to the suggested protocol, i.e. the choice of the restriction enzyme could likely increase the efficiency of the method.
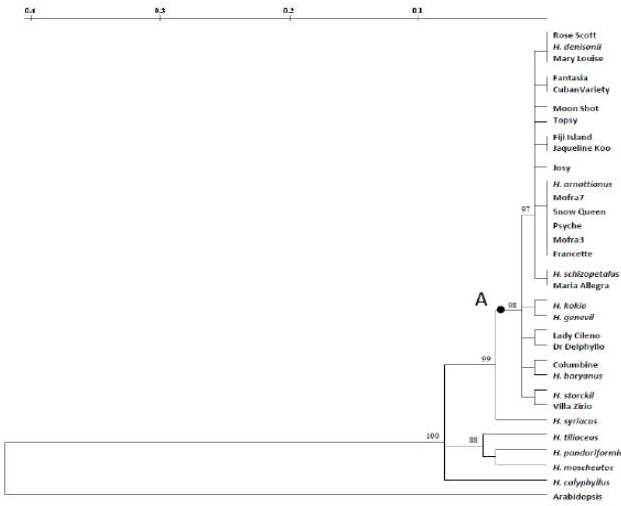
The high conservation sequence degree observed among commercial varieties and the Hibiscus species sexually compatible with H. rosa-sinensis are in agreement with the cytogenetic evidence produced by Singh and Khoshoo (1989), which showed that these inter-fertile species have contributed to the extensive genetic variability of H. rosa-sinensis. The revealed HibMAX2-like sequence analysis is consistent with secondary ranks of taxa (Sections) proposed by Pfeil and Crisp (2005) through chloroplast DNA analysis. In fact, the highest similarity values for the target sequence were achieved among the analyzed H. rosa-sinensis cultivars and the sexually compatible species, all belonging to the Lilibiscus Section, while the lower values were observed for species of different taxonomic Sections such as H. syriacus, H. panduriformis, H. moscheutos, H. tiliaceus and H. calyphyllus. Concerning the H. cannabinus, it could likely either do not possess the HibMAX2 gene, or possess a highly differentiated element, therefore not amplified. Kenaf is one of the fast-growing plants classified in the Furcaria Section of Hibiscus; it has both annual and biennial genotypes, often not branched. This differentiates from the other examined species (shrubs or small trees) characterized by perennial life cycle, with complex vegetative morphologies (Craven et al 2003). Moreover, a previous study (Braglia et al 2010) had revealed the lowest genetic similarity value between H. rosa- sinensis cultivars and kenaf defining this latter as the most distantly related species within the Hibiscus genus.
The occurrence of LRRs in the Hibiscus isolated fragment assigns this sequence among the F-box genes, one of the largest multigene superfamilies involved in shoot lateral branching growth. Members of this protein family function as subunits of the multiprotein Skp-Cullin-Fbox for polyubiquitination and degradation by the 26S proteasome (Xu et al 2009). In particular, the F-box LRR proteins confer substrate specificity to the SCF complex via their two distinct functional domains: the first domain (F-box) binds to another subunit of the SCF complex, the second domain (LRR repeats) interacts with specific proteins to be polyubitquinated (Stirnberg et al 2007).
Although the present results are the first step in the isolation of the whole Hibiscus specific element for MAX2 gene, the cloned fragment can be already investigated for association to the branched trait, to evaluate its utility in marker-assisted breeding schemes. Further studies including the isolation of a cDNA fragment (working back to the full length through RACE-PCR technique), the mRNA expression analysis and the functional variant identification are in turn necessary to better characterize the HibMAX2-like sequence, as well as to clarify its involvement in the axillary branch proliferation mechanisms.
Acknowledgements
We wish to thank Mr. Cesare Bianchini and Dr. Marco Ballardini for their support in managing the germplasm collection. Research founded by the Italian Ministry of Agriculture in the framework of the project “Risorse tecniche e genetiche per il florovivaismo (FLORIS)”.
References
Arite, T., Umehara, M., Ishikawa, S., Hanada, A., Maekawa, M., Yamaguchi, S. & Kyozuka, J. (2009). “D14, a Strigolactone-Insensitive Mutant of Rice, Shows an Accelerated Outgrowth of Tillers,” Plant & Cell Physiology, 50 1416—1424.
Publisher – Google Scholar
Bennett, T., Sieberer, T., Willett, B., Booker, J., Luschnig, C. & Leyser, O. (2006). “The Arabidopsis MAX Pathway Controls Shoot Branching by Regulating Auxin Transport,” Current Biology, 16 553—563.
Publisher – Google Scholar
Booker, J., Sieberer, T., Wright, W., Williamson, L., Willett, B., Stirnberg, P., Turnbull, C., Srinivasan, M., Goddard, P. & Leyser, O. (2005). “MAX1 Encodes a Cytochrome P450 Family Member that Acts Downstream of MAX3/4 to Produce a Carotenoid-Derived Branch-Inhibiting Hormone,” Developmental Cell, 8 443—449.
Publisher – Google Scholar
Braglia, L., Bruna, S., Lanteri, S., Mercuri, A. & Portis, E. (2010) “An AFLP-based assessment of the genetic Diversity within Hibiscus rosa-sinensis and its Place within the Hibiscus Genus Complex,” Scientia Horticulturae, 123 (3) 372-378.
Publisher – Google Scholar
Craven, L. A., Wilson, F. D. & Fryxell, P. A. (2003). “A Taxonomic Review of Hibiscus Sect. Furcaria (Malvaceae) in Western Australia and the Northern Territory,” Australian Systematic Botany, 16 (2) 185-218.
Publisher – Google Scholar
Hillis, D. M. & Bull, J. J. (1993). “An Empirical Test of Bootstrapping as a Method for Assessing Confidence in Phylogenetic Analysis,” Systematic Biology, 42 182-192.
Publisher – Google Scholar
Johnson, X., Brcich, T., Dun, E. A., Goussot, M., Haurogné, K., Beveridge, C. A. & Rameau, C. (2006). “Branching Genes are Conserved Across Species. Genes Controlling a Novel Signal in Pea are Co Regulated by Other Long-Distance Signals,” Plant Physiology, 142 1014—1026.
Publisher – Google Scholar
Leyser, O. (2009). “The Control of Shoot Branching: An Example of Plant Information Processing,” Plant, Cell &Environment 32, 694—703.
Publisher – Google Scholar
Liang, J., Zhao, L., Challis, R. & Leyser, O. (2010). “Strigolactone Regulation of Shoot Branching in Chrysanthemum (Dendranthema Grandiflorum),” Journal of Experimental Botany 61(11), 3069-3078.
Publisher – Google Scholar
Lumba, S., Cutler, S. & McCourt, P. (2010). “Plant Nuclear Hormone Receptors: A Role for Small Molecules in Protein-Protein Interactions,” Annual Review of Cell and Developmental Biology 26, 445—69.
Publisher – Google Scholar
McSteen, P. & Leyser, O. (2005). “Shoot Branching,” Annual Review of Plant Biology 56, 353—374.
Publisher – Google Scholar
Morris, S. E., Turnbull, C. G. N., Murfet, I. C. & Beveridge, C. A. (2001). “Mutational Analysis of Branching in Pea. Evidence that Rms1 and Rms5 Regulate the Same Novel Signal,” Plant Physiology 126, 1205-1213.
Publisher – Google Scholar
Pfeil, B. E. & Crisp, M. D. (2005). “What to Do with Hibiscus? A Proposed Nomenclatural Resolution for a Large and Well Known Genus of Malvaceae and Comments on Paraphyly,” Australian Systematic Botany 18(1), 49-60.
Publisher – Google Scholar
Singh, F. & Khoshoo, T. N. (1989). ‘Cytogenetic Basis of Evolution in Garden Hibiscus,’ Nucleus 32, 62—67.
Google Scholar
Snowden, K. C. & Napoli, C. A. (2003). “A Quantitative Study of Lateral Branching in Petunia,” Functional Plant Biology30, 987-994.
Publisher – Google Scholar
Stirnberg, P., Furner, I. J. & Leyser, H. M. O. (2007). “MAX2 Participates in an SCF Complex which Acts Locally at the Node to Suppress Shoot Branching,” The Plant Journal 50, 80—94.
Publisher – Google Scholar
Stirnberg, P., van den Sande, K. & Leyser, H. M. O. (2002). “MAX1 and MAX2 Control Shoot Lateral Branching in Arabidopsis,” Development 129, 1131—1141.
Publisher – Google Scholar
Van de Peer, Y. & De Wachter, R. (1994). “Treecon for Windows: A Software Package for the Construction and Drawing of Evolutionary Trees for the Microsoft Windows Environment,” Computer Applications in the Biosciences 10, 569—570.
Publisher – Google Scholar
Vos, P., Hogers, R., Bleeker, M., Reijans, M., van de Lee, T., Homes, M., Freiters, A., Pot, J., Peleman, J., Kuiper, M. & Zabeau, M. (1995). “AFLP: A New Technique for DNA Fingerprinting,” Nucleic Acids Research 23, 4407-4414.
Publisher – Google Scholar
Wang, Y., Sun, S., Zhu, W., Jia, K., Yang, H. & Wang, X. (2003). ‘Strigolactone/MAX2-Induced Degradation of Brassinosteroid Branscriptional Effector BES1 Regulates Shoot Branching Developmental,’ Cell 27, 681—688.
Xu, G., Ma, H., Nei, M. & Kong, H. (2009). “Evolution of F-Box Genes in Plants: Different Modes of Sequence Divergence and Their Relationships with Functional Diversification,” Proceedings of the National Academy of Sciencesof the United States of America 106, 835—840.
Publisher – Google Scholar