Introduction
An impairment of the hypothalamic-pituitary-adrenal axis which manifests by an increase in the concentration of cortisole in the blood plasma and brain occurs in the patients with endocrine and neuropsychiatric deseases and is attended by morphologic and functional injuries of brain [1, 2]. It was established that increased levels of glucocorticoids in the blood and the brain caused by chronic stress or the administration of exogenous corticosteroids induce a decrease in the number of pyramidal neurons in the hippocampal area CA3 [3, 4]. In vitro studies revealed a toxic effect of glucocorticoids on cultured hippocampal neurons of rodents [5].
It was also found that various glucocorticoids have site-specific effects on some regions of brain. Corticosterone as nonselective agonist of the glucocorticoid and mineralocorticoid receptors causes the damage of the CA3 pyramidal neurons of the hippocampus [4], while systemic administration of the selective agonist of glucocorticoid receptors dexamethasone causes damage of the dentate gyrus granule cells [6]. However, studies, carried out using more subtle methods, found that systemic administration of dexamethasone in rats caused a dose-dependent activation of microglia (a marker of neuronal apoptosis), an increase in the number of neurons with morphological features of apoptosis and a decrease in immunoreactivity to MAP-2 (a protein of the cytoskeleton of neurons) in the dorsomedial parts of the striatum, the dentate gyrus and areas CA1 and CA3 of the hippocampus, but not in the lateral septal nucleus. These morphological changes were prevented by competitive blocker of glucocorticoid receptors mifepristone [7]. It can be assumed that the observed in various psychiatric illnesses reduction of hippocampal and striatum volume, revealed in the postmortem brain specimens of patients [8, 9], at least partly is a result of impairment of the hypothalamic-pituitary-adrenal axis and increased glucocorticoid concentration in blood plasma and brain.
At the same time it still unclear whether the observed morphological changes in the studied brain structures caused by the impact of the glucocorticoids satisfy with changes in their functional state in the first place in electrophysiological characteristics, especially taking into account the compensatory potential of neurons. In addition, the impact of injury of the neuronal structures evoked by glucocorticoids on its electrophysiological parameters remains unclear.
In present study it was attempted to clarify the mechanisms of functional changes in neuronal populations of the dentate gyrus and area CA1 of the hippocampus induced by selective agonist of the glucocorticoid receptors dexamethasone. One of the main functions of neuron is generation of the action potentials after synaptic depolarization (excitatory postsynaptic potential). Therefore the functional disorders of hippocampal neurons produced by dexamethasone we studied as changes in the efficacy of excitatory glutamatergic synaptic transmission in dentate gyrus and area CA1 of hippocampus. We studied also the impact of chronic administration of imipramine on produced by dexamethasone injuries of the hippocampal neurons so as to ascertain probable neuro- and cerebroprotective action of imipramine in environment of action on an organism of high doses of the corticosteroids.
Research Design and Methodology
Animals
Study was performed on both sexes of Wistar rats (weight 100 — 150 g; the nursery of Kiev Bogomoletz Institute of Physiology of National Academy of Sciences of Ukraine), which were housed in a 12-hour daily regime (12 h light/dark cycle at 8:00 A.M.; 100 lux), by groups of 4-5 animals in a cage, with free access to water and food. All protocols were approved by Ethics Comitte of the Maxim Gorky Donetsk National Medical University.
Electrophysiological Recordings in Slices
The main study was performed on the hippocampal slices [10], which were taken from the animals that were injected intraperitoneally with dexamethasone (experimental group, n = 56) or saline (control group, n = 24) for 24 hours before electrophysiological studies. Decapitation was carried out under ketamine anesthesia (50 mg / kg, intraperitoneally). The brain was isolated from the skull and immediately placed in cooled (4 — 6 º C) solution with reduced concentration of Na+ previously saturated with a gas mixture of 95% O2 and 5% CO2 for 2-3 min. Then, the posterior pole of the brain was fixed on platform of vibratome (The Vibratome Company, St Louis, MO, USA) with cyanoacrylate glue and the transverse slices of dorsal hippocampus (400 μm thickness) were prepared in the bath of vibratome filled with cold solution contained ( in mM): sucrose – 248, NaHCO3- 26, KCl – 3, KH2PO4 – 1.25, CaCl2 – 0.1, MgSO4 – 3, glucose — 10 saturated with gas mixture of 95% O2 and 5% CO2. Then slices of the hippocampus placed in a humidified interface-type holding chamber for at least 1 h before recordings where its were superfused with bicarbonate Krebs solution contained (in mM): NaCl – 124, NaHCO3 – 26, KCl – 3, KH2PO4 – 1.25, CaCl2 – 2, MgSO4 – 1, glucose – 10. The solution in the holding chamber continuously bubbled with 95 % O2 and 5 % CO2; flow rate of the solution – 2 ml / min. The temperature was maintained at 25 ° C, pH of aerated Krebs solution – 7.4. After 60-90 minutes of exposure in holding chamber hippocampal slices were transferred into the working chamber (its volume was 0.5 ml), were fixed by stimulation electrode and were superfused (2 ml/min) with Krebs solution continuously bubbled with 95 % O2 and 5 % CO2. The temperature in the working chamber was maintained at 28 ° C.
Field (f) EPSP of proximal dendrites of pyramidal neurons in area CA1 of the hippocampus (str. radiatum) and dentate gyrus granule cells (proximal 1/3 of str. moleculare) were recorded extracellularly through glass micropippetes filled with a solution of NaCl (2 M) with tip resistance 1 — 3 MΩ. Electrical signals (fEPSP) of studied neurons were recorded using the UBF-4 amplifier (Byophyspribor, Pushchino, RF) and Digidata1322A (Molecular Devices, Union City, CA, USA). Obtained data were stored and analyzed with pCLAMP software (version 6.0.4; Molecular Devices).
Schaeffer collaterals and fibres of perforant path were electrically stimulated using a bipolar nicrome electrode (resistance on dc 200 KΩ) placed in the str. radiatum of area CA1 and middle third of str. moleculare dentate gyrus. Stimulation impulses of constant current (0,02 — 0,08 mA) were generated by ESU 2 stimulator (Electron, Lvov, UA) and delivered through isolation unit every 10 s. For the dynamic characteristics of the efficiency of synaptic transmission, we explored the dependence of the amplitude of focal potentials on the intensity of presynaptic stimulation. At first it was defined the threshold of the postsynaptic response, and then recorded alterations in it amplitude during increasing in the intensity of stimulation by step 5 V. The synaptic reactivity of the dentate gyrus granule neurons and CA1 pyramidal neurons was represented by the ratio of the amplitude of postsynaptic responses in mV to the intensity of presynaptic stimulation in V and expressed as mV/V. The neuronal damage in area CA1 and dentate gyrus were evaluated by reducing of synaptic reactivity.
It was investigated the effect of toxic doses – 7 and 20 mg / kg — of dexamethasone (KRKA, Slovenia, solution in ampulas) in 24 hours after its intraperitoneal administration in rats [7] on efficacy of synaptic transmission in the hippocampus. Equaly volume of saline was administrated to control rats. Components of dexamethasone neurotoxicity associated with activation of neuronal NMDA glutamate receptors and inhibition of expression of the neurotrophins and other factors, which produced the phosporilation of tyrosine residue in neuronal substrates [11, 12] were investigated with the substances-analyzers: non-unit-selective non-competitive NMDA receptor blocker – ketamine (Biolek, Ukraine, solution in ampulas), co-agonist of NMDA receptor – glycine (Reahim, Latvia, powder), an inhibitor of tyrosine phosphoprotein phosphatases – sodium ortovadanate (Uralkhimreaktiv, RF, powder). Three last substances were administrated subcutaneously at doses 30, 50 and 15 mg/kg respectively. We investigated the effects of acute and chronic administration of imipramine (EGIS, Hungary, solution in ampulas) – an inhibitor of norepinephrine and serotonin transporters — on dexamethasone neurotpxicity. In the first case imipramine 25 mg / kg was administered intraperitoneally simultaneously with dexamethasone and 24 hours later slices of the hippocampus were prepared for electrophysiological study. In the second case, imipramine was administered in animals intraperitoneally for 14 days. On the fourteenth day, in addition to imipramine, rats were administered dexamethasone and 24 hours later brain slices were prepared for electrophysiological studies.
Statistical Analysis
Each series of experiments was carried out for 8 – 10 brain slices taken from different 3 — 4 animals. The statistical significance of differences among data groups was assessed using one-way measure ANOVA with Bonferroni/Dunn post hoc analysis or using the two-tailed Student`s t test. The research results were subjected to conventional processing methods of variation statistics using the software package Medstat. The statistical difference of compared values was significant if P < 0,05.
Results and Discussion
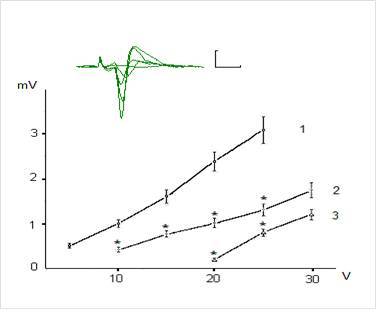
Figures 1 and 2 show examples of field (f)EPSP pyramidal neurons of CA1 and dentate gyrus granule cells and dependence of the fEPSP amplitude on the intensity of presynaptic stimulation. The dependence of fEPSP amplitude of CA1 pyramidal neurons and granule neurons of the dentate gyrus on the intensity of the presynaptic stimulation in the range of 5-25 V has had largely linear character and the slope of the obtained curves was different for CA1 pyramidal and dentate gyrus granule neurons (Fig. 1 and 2).
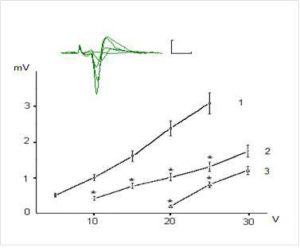
Figure 1. Dependence of Field Epsps Amplitude of the Pyramidal Neurons of CA1 Region on Theintensity of Presynaptic Stimulation in Intact and Exposed to Toxic Doses of Dexamethasone Rats. The Top of the Figure Shows Oscillograms of the Field EPSP Induced by Stimulation of Chaffer Collaterals with Increasing Intensity in the Range of 5-30 V in One of the Hippocampal Slices (Control). Each Depicted Fepsp Averaged Over 10 Realizations. Calibration: 1 Mv, 10 Ms. The Bottom of the Figure Shows Dependence of the EPSP Amplitude on Intensity of Presynaptic Stimulation 10 Intact Rats (1) and in 8 Animals Exposed to Dexamethasons at the Doses of 7 and 20 Mg/Kg (2 and 3, Respectively). The Vertical Scale — The Amplitude of the Field EPSP (Mv), the Horizontal Scale — The Intensity of Presynaptic Stimulation (V). Results Are Expressed as Mean = SEM; *- P<0, 05 Compared with Control
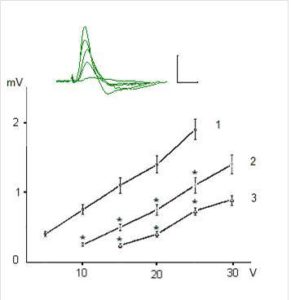
Figure 2. Dependence of Amplitudw of Field Epsps of Dentate Gyrus Granule Neurons on the Intensity of Presynaptic Stimulation in Intact and Exposed to Toxic Doses of Dexamethasone Rats. Notes: See on Fig. 1
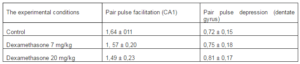
It was revealed that preliminary administration of the toxic doses of dexamethasone induced significant decreasing of the amplitudes of fEPSPs. Indeed, dexamethasone at doses 7 and 20 mg/kg reduced an amplitude of fEPSP of the pyramidal neurons of area CA1 evoked by submaximal (25 V) presynaptic stimulation (Fig. 1) to 1, 24 ± 0,16 and 0,52 ± 0,11 mV respectively vs 2,98 ± 0,30 mV in control (P < 0,05). Likewise, dexamethasone at doses 7 and 20 mg/kg reduced an amplitude of fEPSP of granule cells of the dentate gyrus (Fig. 2) to 0,83 ± 0,12 and 0,51 ± 0,07 mV vs 1,92 ± 0,15 mV in control (P < 0,05). Decrease of amplitudes of fEPSPs of the pyramidal and granule neurons did not accompanied by alterations of the values of pair pulse facilitation and depression (tabl.). This indicates that decrease of the amplitudes of fEPSPs of the pyramidal and granule neurons produced by dexamethasone have postsynaptic nature [13].
Table 1. The Values of Pair Pulse Facilitation and Depression (Interstimulus Interval 50 Ms) Under Different Experimental Conditions
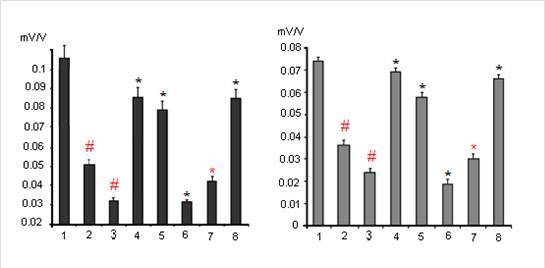
One from early manifestations of dexamethasone neurotoxicity is a decrease of the values of the postsynaptic potentials, i. e. decreasing of a reactivity of the glutamatergic synapses of hippocampal neurons (Fig. 3, columns 2 and 3). Indeed dexamethasone at doses 7 and 20 mg/kg decreased synaptic reactivity of the CA1 pyramidal neurons to 0,052 ± 0,005 mV/V and 0,033 ± 0,003 mV/V respectively vs 0,105 ± 0,012 mV/V in control (P < 0,05) and the dentate gyrus granule neurons to 0,036 ± 0,004 mV/V and 0,024 ± 0,003 mV/V respectively vs 0,074 ± 0,004 mV/V in control (P < 0,05).
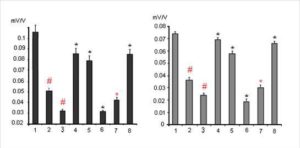
Figure 3.The Changes of Synaptic Reactivity of Pyramidal Neurons of Area CA1 (Left) and Granule Neurons of Dentate Gyrus (Right) Induced by Dexamethasone Toxic Doses and Its Modulation by Substances for Analysis and Imipramine. Columns: 1- Control; 2 and 3 — Effects of Dexamethasone Administrated at the Doses of 7 and 20 Mg/Kg Respectively (N = 10 ); Further Dexamethasone Administrated at the Dose 7 Mg/Kg Against the Background Administration of 30 Mg/Kg Of Ketamine — 4; 15 Mg/Kg of Sodium Orthovanadate — 5; 50 Mg/Kg of Glycine — 6; 25 Mg/Kg Imipramine ( Single Injection — 7 or Chronic Treatment During Two Weeks — 8) ( N = 8 ). The Vertical Scale — Synaptic Reactivity (Mv/V). Results Are Expressed as Mean = SEM; Error Bars Indicate + SEM. # – P < 0, 05 Compared with Control; * – P <0, 05 Compared Dexamethasone in the Dose of 7 Mg/Kg
The inhibiting effect of dexamethasone on glutamatergic synaptic transmission can be the result of the apoptosis of pyramidal and granule neurons in the hippocampus or the destruction of the cytoskeleton in these neurons that were revealed by morphological studies [7]. However, the number of neuronal apoptosis of CA1 and dentate gyrus when exposed to dexamethasone at doses of 7 and 20 mg / kg was limited and almost equal, while the reduction in immunoreactivity to cytosceleton protein MAP-2 was more pronounced when using dexamethasone at the dose of 20 mg / kg [7]. It appears that the observed decrease in reactivity of glutamatergic synapses of hippocampal neurons occures to the disruption of the structure of the cytoskeleton of neurons. Since the values of pair pulse facilitation (pyramidal neurons) and pair pulse depression (granule neurons) in hippocampal slices exposed to dexamethasone did not differ from those of intact slices (tabl.), more probably that dexamethasone neurotoxicity was associated with disruption of the structure of the cytoskeleton of postsynaptic neurons including its the postsynaptic apparatus.
At the same time it is unclear in which way dexamethasone induces damage of the cytoskeleton in neurons. The observed effect may be result from the direct action of dexamethasone on the transcription and sequent translation of cytoskeletal proteins, but may also be mediated by the influence of dexamethasone on calcium homeostasis in neurons or on the expression of different brain neurotrophic factors necessary for viability of the neurons. It was found that dexamethasone at the dose of 7 mg / kg in case of the prior twice administration with 6 h interval of non-competitive NMDA receptor blocker ketamine at the dose of 30 mg / kg subcutaneously exerted a less pronounced inhibitory effect on the synaptic responsiveness of pyramidal (0,085 ± 0,008 mV/V vs 0,052 ± 0,005 mV/V; P < 0,05) and granule (0,069 ± 0,004 mV/V vs 0,036 ± 0,004 mV/V; P < 0,05) neurons (Fig. 3, column 4). On the other hand, (Fig. 3, column 6) inhibition of the synaptic reactivity of the studied neurons induced by 7 mg / kg of dexamethasone was increased after the administration in rats of NMDA glutamate receptor co-agonist glycine at the dose of 50 mg / kg intraperitoneally via 6 h after administration of dexametasone at dose 7 mg/kg (CA1 – 0,033 ± 0,003 mV/V vs 0,052 ± 0,005 mV/V; P < 0,05; dentate gyrus — 0,019 ± 0,002 mV/V vs 0,036 ± 0,004 mV/V; P < 0,05).
In case of combined administration of a single dose of clinically active antidepressant imipramine (25 mg/kg) and glucocorticoid dexamethasone (7 mg/kg) the neurotoxic effects of dexamethasone were increased (Fig. 3, column 7); thus inhibition of synaptic reactivity in CA1 and dentate gyrus increased to 0,040 ± 0,003 mV/V and 0,026 ± 0,002 mV/V respectively under dexamethasone + imipramine vs 0,052 ± 0,005 mV/V and 0,036 ± 0,004 mV/V respectively under dexamethasone only ( P = 0,05). However, chronic administration within 2 weeks of imipramine in rats has the opposite effect, namely, the decreasing dexamethasone neurotoxicity, manifested significantly smaller decrease in synaptic reactivity in rats induced by 7 mg / kg of dexamethasone (Fig. 3, column 8) — 0,086 ± 0,008 and 0,064 ± 0,004 mV/V vs 0,052 ± 0,005 and 0,036 ± 0,004 mV/V for neurons CA1 and dentate gyrus respectively (P < 0,06). Likewise chronic administration of imipramine acute aministration of 15 mg/kg of sodium orthovanadate (Fig. 3, column 5) decreased an inhibition of synaptic reactivity of CA1 and dentate gyrus neurons – 0,078 ± 0,007 and 0,060 ± 0,003 mV/V vs 0,052 ± 0,005 and 0,036 ± 0,004 mV/V respectively (P < 0,05).
The neurotoxic effects of dexamethasone manifesting by inhibition of synaptic reactivity of the studied neurons may be partially mediated by activation of NMDA glutamate receptors. It was found that glucocorticoids inhibit glucose uptake by both neurons and glial cells that results in the decreasing of energy production in these cells. Deficit of energy causes a decrease of activity of transporters, including synaptic neuronal and glial glutamate transporters [14,15]. The decreasing of glutamate transporters activity leads to leakage of glutamate out of synapses. Increasing the concentration of glutamate in the extracellular spaces of the brain adduces to the activation of synaptic and extrasynaptic high affinity the NMDA glutamate receptors. The ion channels of the NMDA receptors have a high calcium permeability [16]. This is the reason for aberration of calcium homeostasis, accompanied by a rise of cytoplasmic Ca2+ concentration with subsequent activation of Ca2+-dependent proteases and phospholipases and the destruction of the cytoskeleton in neuronal dendrites. The decreasing of the neurotoxic effects of glucocorticoids by ketamine (Fig. 3, column 4) and, on the other hand, amplifing of glucocorticoid neurotoxicity by glycine in the dose of 50 mg / kg (Fig. 3, column 6) argue about fairness this assumption, whereas the NMDA antagonists abate but the co-agonists of the NMDA receptors reinforce glutamate neurotoxicity [17, 18]. It was also found that chronic administration of antidepressant drug impairs the function of neuronal NMDA receptors. This is manifested by the inhibition of mRNA NMDA subunit receptors formation in the hippocampus and decreasing of the amplitude of NMDA-components of EPSP of dentate gyrus neurons [19,20]. Therefore chronic administration of imipramine may reduce glutamatergic component glucocorticoid neurotoxicity.
Reinforcing by single injection of imipramine of neurotoxic action of dexamethasone (Fig. 3, column 7) probable is conditioned by raising of the level of corticosteroid in the cytoplasm of pyramidal and granule neurons. Indeed, studies on cultured neurons and leukocytes found that different antidepressants — monoamine re-uptake inhibitors — decrease activity of membrane verapamil-sensitive transporter which removes corticosteroids from the internal environment of cells [18,21]. On the other hand, it is known that chronic administration of antidepressants of different functional classes accompanied by increased activity of signaling pathway adenylyl cyclase – cAMP – protein kinase A. This, in turn, leads to increased level of phosphorylation of Ca2+ / cAMP-dependent transcription factor CREB, which reinforces transcriptional activity in the neuronal nucleus. It exists conclusive data that chronic administration of different antidepressants produced reinforcement of activity of the mitogen-activated protein kinases (ERK 1/2), and expression of the neurotrophins and growth factors (BDNF, NT-3, VEGF) ets., which enhance the phosphorilation of neuronal substrates by tyrosine kinases and possess powerful neuroprotective action [22 —24]. Besides, it is known that glucocorticoids, as stress hormones, inhibit activity of a series of the protein kinases and expression of the neurotrophins and growth factors [25]. These facts allow to think that chronic administration of imipramine contributes the reinforcement of phosphorilation of the cytosceleton proteins by the tyrosine kinases and stabilizes the neuronal cytosceleton.
The level of tyrosine phosphorylation of synaptic proteins in addition to activity of the tyrosine kinases is also regulating by tyrosine phosphoprotein phosphatases whose activity is inhibited by arsenate and vanadate [26]. Decreasing of glucocorticoid neurotoxicity by sodium orthovanadate (Fig. 3, column 5) indicates that the degree of dephosphorylation of tyrosine residues in the structure of synaptic proteins is reduced by the sodium orthovanadate that in turn, increases the level of its phosphorylation, despite the reduction of activity of the protein kinases, expression of the neurotrophins and growth factors caused by the impact of dexamethasone.
Conclusions
The excessive dose of glucocorticoids induces functional and morphological impairments of the cortical and hippocampal neurons. In the current study it was shown that early manifestation of neurotoxic action of synthetic glucocorticoid dexamethasone is inhibition of synaptic reactivity of the pyramidal neurons of area CA1 hippocampus and the granule neurons of dentate gyrus. The neurotoxic action of dexamethasone probably is conditioned by increase of functional activity of the NMDA receptors and by decrease of tyrosine kinase phosphorylation of neuronal substrates produced by the protein kinases, neurotrophins and growth factors in researched neurons. The chronic administration of imipramine reduced the neurotoxic action of dexamethasone probably by the attenuation of NMDA glutamate receptor activity and increasing of tyrosine kinase phosphorylation of the neuronal substrates. Thus imipramine and possibly others antidepressants might to complement a list of the neuro- and cerebroprotectors.
Conflict of Interest
The authors declare no conflict of interest
Acknowledgments
We thank Dr. Andrey Savustyanenko and Dr. George Adu-Gyamfi for helpful comments and discussion during preparation of the manuscript.
References
1.McEwen, B. S. (2007). “Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain,”Physiological Reviews, 87 (3) 873—904.
Publisher – Google Scholar
2. De Kloet, E. R., Joëls, M. & Holsboer, F. (2005). “Stress and the Brain: From Adaptation to Disease,” Nature Review of Neuroscience, 6 (6) 463—475.
Publisher – Google Scholar
3. Galea, L. A. M., Mcewen, B. S., Tanapat, S., Deak, T., Spencer, R. L. & Dhabhar, F. S. (1997). “Sex Differences in Dendritic Atrophy of CA3 Pyramidal Neurons in Response to Chronic Restraint Stress,” Neuroscience 81, (3) 689—697.
Publisher – Google Scholar
4. Reagan, L. P. & Mcewen, B. S. (1997). “Controversies Surrounding Glucocorticoid-Mediated Cell Death in the Hippocampus,” Journal of Chemical Neuroanatomy, 13 (2) 149—167.
Publisher – Google Scholar
5. Mcintosh, L. J. & Sapolsky, R. M. (1996). “Glucocorticoids Increase the Accumulation of Reactive Oxygen Species and Enhance Adriamycin-Induced Toxicity in Neuronal Culture,” Experimental Neuroogy, 141 (2) 201—206.
Publisher – Google Scholar
6. Sousa, N., Paulabarbosa, M. M. & Almeida, O. F. X. (1999). “Ligand and Subfield Specificity Corticoid-Induced Neuronal Loss in the Hippocampal Formation,” Neuroscience, 89 (4) 1079—1087.
Publisher – Google Scholar
7. Haynes, L. E., Griffiths, M. R., Hyde, R. E., Barber, D. J. & Mitchell, I. J. (2001). “Dexamethasone Induces Limited Apoptosis and Extensive Sublethal Damage to Specific Subregion of the Striatum and Hippocampus: Implication for Mood Disorders,” Neuroscience, 104 (1) 56—79.
Publisher – Google Scholar
8. Bremner, J. D., Narayan, M., Anderson, E. R., Bronen, R. A., Seibil, J. P., Southwick, S. M., Delaney, R. C., Mccarthy, G., Charney, D. S. & Innis, R. B. (2000). “Hippocampal Volume Reduction in Major Depression,” American Journal of Psychiatry, 157 (1) 115—117.
Publisher – Google Scholar
9. Rogers, M. A., Bradshaw, J. L., Pantelis, C. & Phillips, J. G. (1999). “Frontostriatal Deficits in Unipolar Major Depression,” Brain Research Bulletin, 47 (4) 297—310.
Publisher – Google Scholar
10. Xiao, M.- Y., Karpefors, M., Gustafsson, B. & Wigstrom, H. (1995). “On the Linkage between AMPA and NMDA Receptor Mediated Epsps in Homosynaptic Long-Term Depression in the Hippocampal CA1 Region of Young Rats,”Journal of Neuroscience, 15 (6) 4496 — 4506.
Publisher – Google Scholar
11. Venero, C. & Borrell, J. (1999). “Rapid Glucocorticoid Effects on Excitatory Amino Acid Levels in the Hippocampus: A Microdialysis Study in Freely Moving Rats,” European Journal of Neuroscience, 11 (10) 2465—2473.
Publisher – Google Scholar
12. Schaaf, M. J. M., Dejong, J., Dekloet, E. R. & Vreugdenhil, E. (1998). “Downregulation of BDNF Mrna and Protein in the Rat Hippocampus by Corticosterone,” Brain Research, 813 (1 — 2) 112—120.
Publisher – Google Scholar
13. Zalutsky, R. A. & Nicoll, R. A. (1990). “Comparison of Two Forms Long-Term Potentiation in Single Hippocampal Neurons,” Science 248 (4963) 1619 — 1624.
Publisher – Google Scholar
14. Sapolsky, R. M. (1986). “Glucocorticoid Toxicity in the Hippocampus: Reversal by Supplementation with Brain Fuels,” Journal of Neuroscience, 6 (8) 2240—2244.
Publisher – Google Scholar
15. Virgin, C. E., Ha, T. P., Packan, D. R., Tombaugh, G. C., Yang, S. H., Horner, H. C. & Sapolsky, R. M. (1991). “Glucocorticoids Inhibit Glucose Transport and Glutamate Uptake in Hippocampal Astrocytes: Implication for Glucocorticoid Neurotoxicity,” Journal of Neurochemistry, 57, (4) 1422—1428.
Publisher – Google Scholar
16. Collingridge, G. L. & Singer, W. (1990). “Excitatory Amino Acid Receptors and Synaptic Plasticity,” Trends in Pharmacological Science, 11 (6) 290 — 296.
Publisher – Google Scholar
17. Budd, S. L. & Lipton, S. A. (1999). “Signaling Events in Nmda Receptor-Induced Apoptosis in Cerebrocortical Cultures,” Annals of New York Academy of Science 893 (2) 261 — 264.
Publisher – Google Scholar
18. Zhou, M. & Baudry, N. (2006). “Developmental Changes in NMDA Neurotoxicity Reflect Developmental Changes in Subunit Composition of Nmda Receptors,” Journal of Neuroscience 26 (11) 2956 — 2963.
Publisher – Google Scholar
19. Boyer, P.- A., Skolnick, P. & Fossom, L. H. (1998). “Chronic Administration of Imipramine and Citalopram Alters the Expression of NMDA Receptor Subunit Mrnas in Mouse Brain,” Journal of Molecular Neuroscience, 10 (3) 219—233.
Publisher – Google Scholar
20. Abramets, I. I., Kidin, Y. V., Kuznetsov, Y. V. & Talalayenko, A. N. (2005). “Effects of Behavioral Depression and Chronic Influence of Antidepressants on NMDA/Glutamate Receptor-Mediated Responses of Neurons of the Rat Gyrus Dentatus,” Neurophysiologia 37 (2) 124—133. [Article on Russian].
Publisher
21. Pariante, C. M., Makoff, A. & Levestone, S. (2001). “Antidepressants Enhance Glucocorticoid Receptor Function in Vitro by Modulating the Membrane Steroid Transporters,” British Journal of Pharmacoogy, 134 (6) 1335—1343.
Publisher – Google Scholar
22. Duman, C. H., Schlesinger, L., Kodama, M. Russell, D. S. & Duman, R. S. (2007). “A Role for MAPK Signaling in Behavioral Models of Depression and Antidepressant Treatment,” Biological Psychiatry 61 (5) 661—670.
Publisher – Google Scholar
23. Nibuya, M., Morinobu, S. & Duman, R. S. (1995). “Regulation of BDNF and trkB mRNA in Rat Brain by Chronic Electroconvulsive Seizure and Antidepressant Drug Treatments,” Journal of Neuroscience 15 (11) 7539—7547.
Publisher – Google Scholar
24. Newton, S. S., Colier, E. F., Hunsberger, J., Adams, D., Terwilliger, R., Selvanayagam, E. et al. (2003). “Gene Profile of Electroconvulsive Seizures: Induction of Neurotrophic and Angiogenic Factors,” Journal of Neuroscience 23 (34) 10841—10851.
Publisher – Google Scholar
25. Pittenger, C. & Duman, R. S. (2008). “Stress, Depression, and Neuroplasticity: A Convergence of Mechnisms,”Neuropsychopharmacology 33 (1) 88 109.
Publisher – Google Scholar
26. Lee, S. & Wang, Q. (2007). “Recent Development of Small Molecular Specific Inhibitor of Protein Tyrosine Phosphatase 1B,” Medical Research Review 27 (4) 553 — 573.
Publisher – Google Scholar