Introduction
In addition to antigenic drift (Alexander, 2007; Cox, 2000; Horimoto, 2005), caused by errors in viral replication and the antigenic pressure applied to the surface hemagglutinin and neuraminidase antigens by the immune response, the evolution of influenza virus is shaped by the reassortment process (Kaverin, 2010; Webster, 2004). Influenza virus has a high potential to reassort due to the segmented nature of its genome that consists of eight negative-strand RNA segments. When two different strains of influenza virus coinfect the same cell, progeny viruses (reassortants) are created that contain genes derived from each parent. Genetic reassortment among influenza viruses occurs naturally and plays the important role in viral epidemiology and pathogenicity (Horimoto, 2001).
At least two of the major Influenza pandemics of the 20th century, H2N2 in 1957 and H3N2 in 1968, resulted from reassortments between viruses from two different hosts; avian and human (Forrest, 2010). The reassortment event in 1998 gave rise to the triple reassortant, which is the ancestor of the 2009 pandemic virus (Horimoto, 2001; Forrest, 2010). Subsequent reassortment with H1N1 swine strain resulted in generation of triple reassorted swine A (H1N1) virus that caused the 2009 H1N1 influenza pandemic (Horimoto, 2001; Forrest, 2010). Therefore, study of natural reassortants of the influenza virus is important for epidemiology, diagnosis and vaccination of this infection.
Since late 2005, highly pathogenic avian influenza (HPAI) viruses of H5N1 subtype have spread into Europe, the Middle East and Africa affecting both wild birds and poultry (Lei, 2011). Three distinct sublineages were identified in these regions, termed EMA1 (2.2.1), EMA2 (2.2.2) and EMA3 (2.2.3) by Salzberg et al. (2007) (Salzberg, 2007). Incursions into the European Union have been seen mainly in the wild birds, and the limited outbreaks occurring in poultry, as a result of the spread from wild birds or from poultry to other domestic birds, have been promptly eradicated in the vast majority of the European countries. In contrast, in Africa and in the Middle East, since the first outbreaks reported in early 2006, there has been extensive spreading and circulation of H5N1 virus in poultry, except only an extremely limited number of isolations from wild birds (Horimoto, 2001; Webster, 2004). It is still unclear how H5N1 virus reached these two regions, whether through wild bird movements, trade of infected poultry commodities or both.
The first report of highly pathogenic avian influenza A (H5N1) virus in Kazakhstan was reported for the first time in July 2005 in Pavlodar region (North-East Kazakhstan), and then it was isolated in Akmolinsk region (Central Kazakhstan), North Kazakhstan region and Karaganda region (Central Kazakhstan). In March 21, 2006, a dead wild swan was found in Mangystau region (South-West Kazakhstan) on Caspian Sea coast. Assay of the pathology finding from this bird suggested avian influenza infection of A/H5N1 type. 70 wild birds have died from H5N1 epizooty in this region in total (Tabynov, 2008; Kydyrbaev, 2010). The phylogenetic analysis has shown that the virus which has been registered in Mangystau region was different from EMA groups of HPAI H5N1 viruses and had NS2A genotype that was typical for Gs/Gd-like strains (Chervyakova, 2011; Bogoyavlenskiy, 2011). The study of biological properties has shown that the virus had high pathogenic properties (Sayatov, 2007).
This study is focused on the molecular and phylogenetic analysis of H5N1 virus isolated in Mangystau region of Kazakhstan.
Methods
Virus Isolation and Characterization
Virus was isolated from dead mute swan found on Caspean Sea coast and characterized in the laboratory of ecology of viruses of the Institute of microbiology and virology, Almaty, Kazakhstan and in the Institute for Biological Safety Problems, Ste Gvardeiskiy, Zhambyl Region, Kazakhstan. To isolate the virus, the samples of bird visceral organs were mixed with an equal volume of phosphate buffered saline containing antibiotics (penicillin 2000 U/ml, streptomycin 2 mg/ml and gentamicin 50 μg/ml), incubated for 20 minutes in room temperature, and centrifuged at 1,500 × g for 15 min. The supernatant (0.2 ml/egg) was inoculated into the allantoic route of 9-day-old hen embryonated eggs as described in European Union Council Directive 92/40/EEC [15]. Embryonic death within the first 24 hours of incubation was considered non-specific and these eggs were discarded. After incubation at 37°C for 72 h, the allantoic fluid was collected and tested by haemagglutination (HA) assay as described in European Union Council Directive 92/40/EEC. In the cases where no influenza A virus was detected during initial virus isolation attempt, the allantoic fluid was passaged twice in 9-day-old hen embryonated eggs. The number of virus passages in hen embryonated eggs was limited to the maximum two, to limit laboratory manipulation. A sample was considered negative when the second passage HA test was negative. The subtypes of the virus isolates were determined by conventional haemagglutination inhibition (HI) test and neuraminidase inhibition (NI) test, as described in European Union Council Directive 92/40/EEC.
RNA Extraction and PCR
RNA was extracted from infective allantoic fluid using RNeasy Mini Kit (QIAGENE, GmbH, Germany) accordingly to the manufacturer’s instructions. RNA was converted to full-length cDNA using reverse transcriptase. The RT mix comprised 2.5 μl of DMPC water, 5 μl of 5× First Strand buffer (Invitrogen), 0.5 μl of 10 mM dNTP mix (Amersham Biosciences), 2 μl of 50 mM UNi12 primer, 32 U of RNAguard (Amersham Biosciences), 200 U of MMLV reverse transcriptase (Invitrogen) and 5 μl RNA solution in total volume of 25 μl. The reactions were incubated at 42°C for 60 min, followed by inactivation of the enzyme at 95°C for 5 min.
PCR amplification with different gene specific primers (22 pairs of primers) was performed to amplify the product containing the full length of all genes of influenza. Twenty-five micro liters PCR-mix contained 1× Platinum Taq buffer (Invitrogen), 200 μM dNTP, 2.5 mM MgCl2, 240 nM each of Fw primer and Rw primer, 1 U Platinum Taq DNA Polymerase (Invitrogen) and 3 μl cDNA. Reactions were placed in a thermal cycler at 95°C for 2 min, then cycled 35 times between 95°C 20 sec, annealing at 58°C for 60 sec and elongation at 72°C for 90 sec and were finally kept at 8°C until later use.
Phylogenetic and Sequence Analysis
Sequences of purified PCR products were determined using gene specific primers and BigDye Terminator version 3.1 chemistry (Applied Biosystems, Foster City, CA), according to the manufacturer’s instructions. Reactions were run on ABI310TM DNA analyzer (Applied Biosystems). Sequencing was performed at least twice in each direction. After sequencing, assembly of sequences, removal of low quality sequence data, nucleotide sequence translation into protein sequence, additional multiple sequence alignments and processing were performed with the Bioedit software version 7.0.4.1 with an engine-based on the Clustal W algorithm. The phylogenetic analysis, based on complete gene nucleotide sequences, were conducted using Molecular Evolutionary Genetics Analysis (MEGA, version 5.0) software employing neighbor-joining tree inference analysis with the Tamura-Nei γ-model, with 1000 bootstrap replications to assign confidence levels to branches (Saitou, 1987; Tamura, 2004; Tamura, 2011).
Sequences Obtained from GenBank
The 29 previously described and selected by BLAST (>90% amino acid sequence similarity) sequences of influenza viruses were used for comparison with avian influenza strain isolated in Kazakhstan (Table 1). The choice of strains was carried out according to the following criteria: representatives of different geographical regions; various periods of isolation; and representatives of which are within the average statistical monophyletic groups of strains on the phylogenetic tree. Such strains selection allows to simply assessing the presence or absence of reassortment in influenza virus.
Nucleotide Sequence Accession Numbers
The nucleotide sequences for all viral segments of strain A/mute swan/Aktau/1460/06 analyzed in this study are available from GenBank under accession numbers from FJ434373 to FJ434380.
Results and Discussion
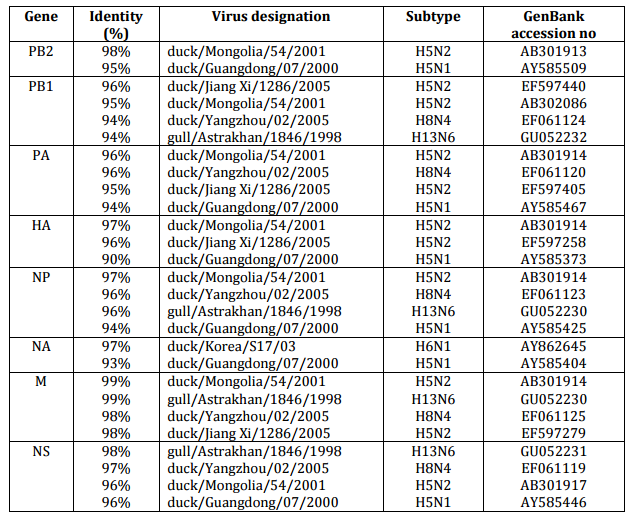
29 sequences of influenza viruses previously described and selected by BLAST (>90% amino acid sequence similarity) were used for comparison with avian influenza strain isolated in Kazakhstan (Table 1). It is interesting that the program had selected various strains of avian influenza for different genes. Thus, the internal virus segments excluding PB2 gene formed a close group with viruses isolated in Yangzhou, Jiang Xi and Astrakhan with high degree of identity (Table 1). Genes PB2 and HA formed a separate branch with strains isolated in Mongolia and Guangdong. The NA gene was distinctly related to the isolates from Korea (Table 1).
Table 1. Influenza A Viruses with Highest Nucleotide Sequence Identity to H5N1 Influenza Virus (A/Mute Swan/Aktau/1460/06) as Determined by BLAST Search in the NCBI.
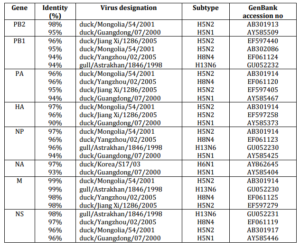
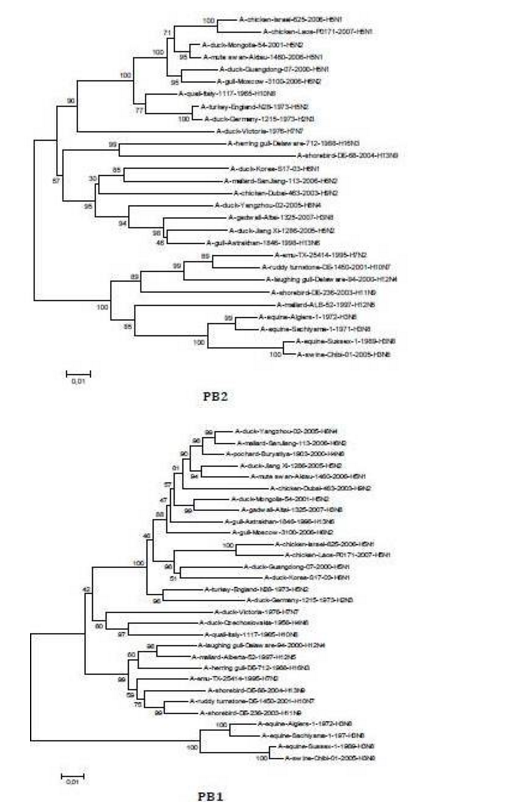
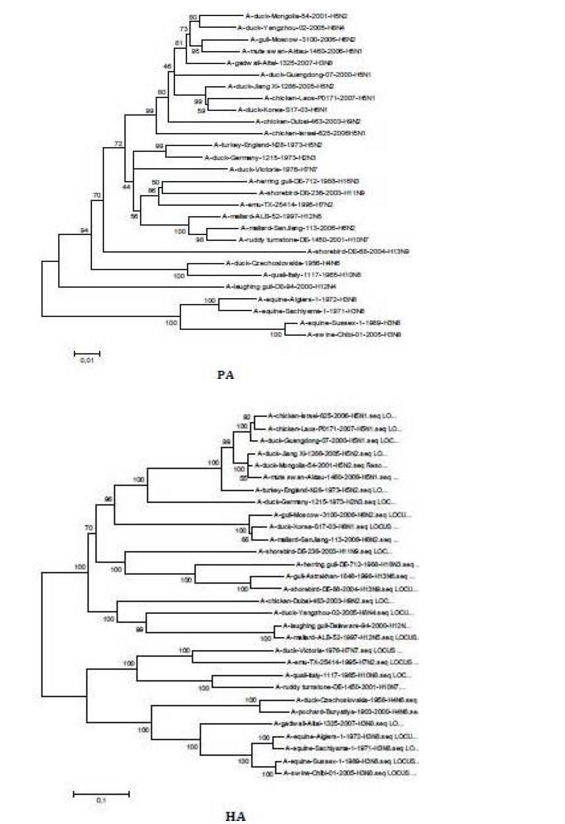
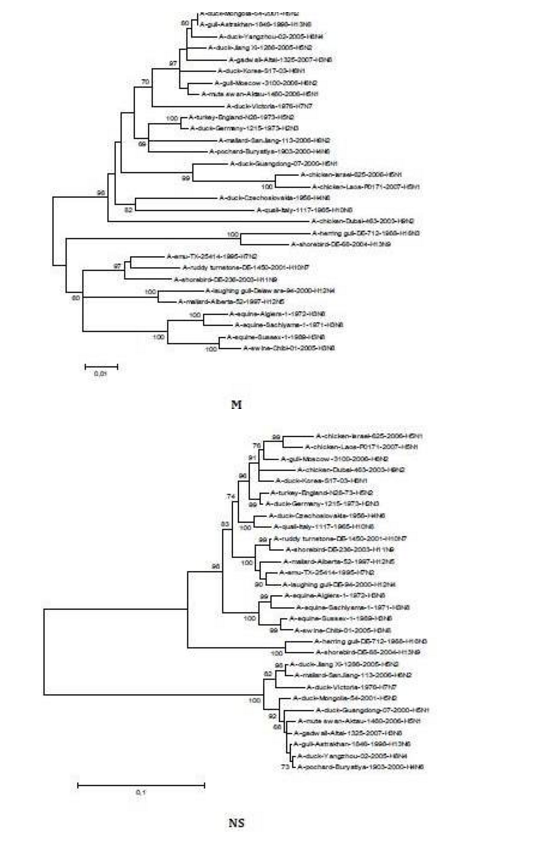
The sequences of all eight gene segments of avian influenza isolate A/mute swan/Aktau/1460/06 were determined and were compared to other sequences that were available in GenBank (Fig. 1). A phylogenetic analysis of eight gene segments showed that the Kazakhstanian isolate was of avian origin and, in general, showed the highest amino acid sequence similarity to avian influenza gene pool of the western Pacific migratory flyway with viruses from China, Korea, Japan, Mongolia and Siberia. However, it was shown that there has also been “mixing” attitudinally through overlapping migratory flyways in the region of Caspian Sea. It was interesting that sequences of PB2, HA and NA genes were formed from some different evolutionary groups.
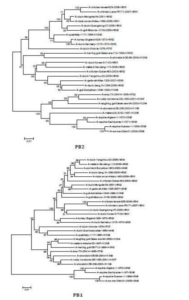
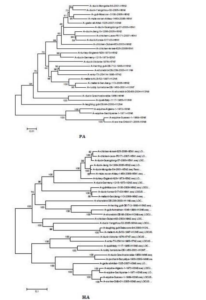
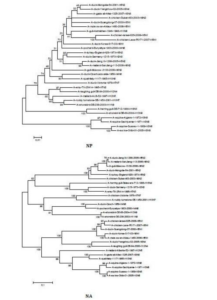
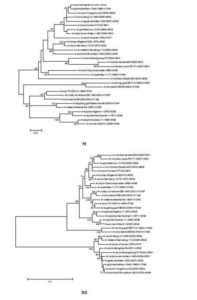
Figure 1 – Phylogenetic Trees of Virus Genes Isolated from Mute Swan in Kazakhstan. Trees were Generated by Using Neighbor-Joining Analysis. The Evolutionary Distances Were Computed Using the Tamura-Nei Method and are in the Units of the Number of Base Substitutions per Site. The Percentage of Replicate Trees in which the Associated Taxa Clustered Together in the Bootstrap Test (1000 Replicates) are Shown Next to the Branches. Analysis was Based on Total Nucleotides 1048 bp of the PB2 Gene, 1194 bp of the PB1 Gene, 1219 bp of the PA Gene, 1559 bp of the HA Gene, 429 bp of the NP Gene, 560 bp of the NA Gene, 961 bp of the M Gene and 800 bp of the NS Gene. All Positions Containing Gaps and Missing Data were Eliminated from the Dataset (Complete Deletion Option). Phylogenetic Analyses were Conducted in MEGA4 [4]. The Mute Swan Genes are Indicated in Box.
Therefore we decided to use the FluGenome, an online set of tools software, for the classification of different lineages and genotypes of influenza virus A/mute swan/Aktau/1460/06 (H5N1) that arose by divergence between different influenza strains cocirculated in the same territory (Lu, 2007).
It was shown that influenza virus A/mute swan/Aktau/1460/06 (H5N1) had the genotype [K,G,D,5H,F,1H,F,2A] and was natural reassortant from different lineages of influenza evolution. The possible derivation of this strain is summarized in Fig. 2. By analysis data of genotyping it has been detected that this new unique genotype arose from reassortment events between three different groups.
Figure 2 – FluGenome Detects the Genesis of Triple Reassortant Influenza A Virus Isolated in Kazakhstan
The origin of the strain A/mute swan/Aktau/1460/06 is unclear. The authors suggest that this H5N1 reassortant virus acquired its PB2, PB1, PA, NP, M and NS gene segments from viruses isolated in China and circulating from 1992 to 2005, whose genotype were [K,G,D,5J,F,1J,F,2A]. These strains have subtypes H5N1, H5N2 and H3N8. The HA genes are descended from A/duck/Mongolia/54/2001 (AB241614), A/duck/Jiang Xi/6146/2003 (EF597255), and A/duck/Gui Yang/3799/2005-like viruses (EF597261). These viruses belong to H5N1, H5N2 and H5N3 subtypes. The remaining NA gene segment might have been originated from a group of viruses which predominantly were isolated in Europe between 1999 and 2005 and have H7N1, H1N1, H6N1 and H11N1 subtypes.
Here, the authors report the results which indicate that the new virus is a triple reassortant, as a result of reassortment event between two geographical lineages. The virus had acquired six inner gene segments from one group, HA gene from another group and NA gene from a third group. This fact confirms that genetically different viruses can cocirculate at the same territory, as previously suggested (Fasina, 2007; Gaidet, 2007; Pittman, 2008).
The present results indicate that a new H5N1 virus with unique genotype was found in mute swan. The similarity of the inner and HA genes with the same of China isolates indicate that they had some connection with Asian lineage of evolution. The similarity of PB2 and NA genes with one of the strains isolated in Europe indicates that the strain A/mute swan/Aktau/1460/06 had a relationship with European lineage of evolution.
Conclusion
This study demonstrates the dynamic nature of the influenza virus genes pool in Eurasia with continuing gene exchanges between the eastern and western parts of the continent. The findings suggest that the H5N1-like virus were likely derived directly from viruses resident in migratory birds. The failure to identify the source for all gene segments of A/mute swan/Aktau/1460/06 virus and its variant highlights the need for continuance of surveillance in both migratory and domestic populations of birds in large regions. Such surveillance is crucial for effective pandemic influenza preparedness.
Acknowledgments
The study was supported by USDA-ISTC partner project (K-747p, Institute of Microbiology and Virology funding, and USDA CRIS (6612-32000-038-00 D)) and with a grant of the Ministry of Science and Education, Republic of Kazakhstan, 0106РК00581.
The authors express their deep appreciation to Sayatov M. and staff of the laboratory ecology of viruses for the opportunity to work with the virus.
References
Alexander, D. J. (2007). “Summary of Avian Influenza Activity in Europe, Asia, Africa, and Australasia, 2002-2006,” Avian Diseases, 51(1 Suppl) 161-166.
Publisher – Google Scholar – British Library Direct
Bogoyavlenskiy, A., Berezin, V., Korotetskiy, I. et al. (2011). ‘Genetic Characterization of H4, H5 and H13 Type Avian Influenza Viruses Isolated from Wild Birdsa in Kazakhstan,’ Influenza and Other Respiratory Viruses, Vol. 5, 60-62.
Chervyakova, O. V., Strochkov, V. M., Sultankulova, K. T., Sandybayev, N. T., Zaitsev, V. L. & Mamadaliyev, S. M. (2011). “Molecular and Genetic analysis of NS Gene from High Pathogenic Strains of the Avian Influenza (H5N1) Virus Isolated in Kazakhstan,” Gene, 476 (1-2) 15-9.
Publisher – Google Scholar
Cox, N. J. & Subbarao, K. (2000). “Global Epidemiology of Influenza: Past and Present,” Annual Review of Medicine, 51 407—421.
Publisher – Google Scholar – British Library Direct
European Union Council Directive 92/40/EEC of 19 May 1992. “Introducing Community Measures for the Control of Avian Influenza,” Official Journal L 167, 22/06/1992. 0001-0016.
Publisher – Google Scholar
Fasina, F. O., Meseko, A. C., Joannis, T. M., Shittu, A. I., Ularamu, H. G., Egbuji, N. A., Sulaiman, L. K. &
Onyekonwu, N. O. (2007). “Control Versus No Control: Options for Avian Influenza H5N1 in Nigeria,” Zoonoses Public Health, Vol. 54(5) 173-176.
Publisher – Google Scholar – British Library Direct
Forrest, H. L. & Webster, R. G. (2010). “Perspectives on Influenza Evolution and the Role of Research, “ Animal Health Research Review, 11(1) 3-18.
Publisher – Google Scholar
Gaidet, N., Dodman, T., Caron, A., Balança, G., Desvaux, S., Goutard, F., Cattoli, G., Lamarque, F., Hagemeijer, W. & Monicat, F. (2007). “Avian Influenza Viruses in Water Birds, Africa,” Emerging Infectious Diseases, Vol. 13(4) 626-629.
Publisher – Google Scholar – British Library Direct
Horimoto, T. & Kawaoka, Y. (2001). “Pandemic Threat Posed by Avian Influenza A Viruses,” Clinical Microbiology Reveiws, 14(1) 129-49.
Publisher – Google Scholar – British Library Direct
Horimoto, T. & Kawaoka, Y. (2005). “Influenza: Lessons from Past Pandemics, Warnings from Current Incidents,” Nature Reviews Microbiology, 3 591—600.
Publisher – Google Scholar – British Library Direct
Kaverin, N. (2010). “Postreassortment Amino Acid Substitutions in Influenza A Viruses,” Future Microbiology, 5(5) 705-15.
Publisher – Google Scholar
Kydyrbayev, Z. K., Tabynov, K. K., Ryskeldynova, S. Zh., Mamadaliyev, S. M. & Khairullin B. M. (2010). “Immunogenicity of the Inactivated Oil Emulsion Influenza A (H5N1) Vaccine in Chickens,” Agriculture and Biology Journal of North America, 1(3) 201-207.
Publisher
Lei, F. & Shi, W. (2011). “Prospective of Genomics in Revealing Transmission, Reassortment and Evolution of Wildlife-Borne Avian Influenza A (H5N1) Viruses,” Current Genomics, 12(7) 466-74.
Publisher – Google Scholar
Lu, G., Rowley, T., Garten, R. & Donis, R. O. (2007). “FluGenome: A Web Tool for Genotyping Influenza A Virus,” Nucleic Acids Research, Vol 35, W275—W279.
Publisher – Google Scholar
Pittman, M. & Laddomada A. (2008). “Legislation for the Control of Avian Influenza in the European Union,” Zoonoses and Public Health, Vol. 55(1) 29-36.
Publisher – Google Scholar – British Library Direct
Saitou, N. & Nei, M. (1987). “The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees,” Molecular Biology and Evolution, 4 406-425.
Publisher – Google Scholar
Salzberg, S. L., Kingsford, C., Cattoli, G., Spiro, D. J., Janies, D. A., Aly, M. M., Brown, I. H., Couacy-Hymann, E., De Mia, G. M., Dung, D. H., Guercio, A., Joannis, T., Ali, A. S. M., Osmani, A., Padalino, I., Saad, N. D., Savić, V., Sengamalay, N. A., Yingst, S. L., Zaborsky, J., Zorman-Rojs, O., Ghedin, E. & Capua, I. (2007). “Genome Analysis Linking Recent European and African Influenza (H5N1) Viruses,” Emerging Infectious Diseases, Vol. 13(5) 713-718.
Publisher – Google Scholar
Sayatov, M. K. h., Kydyrmanov, A., Asanova, S. et al. (2007). ‘Influenza A Virus Surveillance in Wild Birds in Northern and Eastern Caspian in 2002-2006,’ In: International Conference Materials “Preparedness to the Influenza Pandemic — An International Outlook”., Saint—Petersburg, Russia, 2007, 79-80.
Tabynov, K. K., Mambetaliyev, M. A., Mamadaliyev, S. M., Bulatov, Ye. A. & Kopochenya, A. A. (2008). ‘Determination of the Intravenous Pathogenicity Index of the Influenza A Virus Isolated in Kazakhstan in 2005-2006,’ Biotechnology in the republic of Kazakhstan: problems and trends of innovation development, 2008 P. 209-211.
Tamura, K., Nei, M. & Kumar, S. (2004). “Prospects for Inferring Very Large Phylogenies by Using the Neighbor-Joining Method,” Proceeding of the National Academy of Sciences of the United States of America, 101:11030-11035.
Publisher – Google Scholar – British Library Direct
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. & Kumar, S. (2011). “MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods,” Molecular Biology and Evolution, 28(10) 2731-2739.
Publisher – Google Scholar
Webster, R. G. & Hulse, D. J. (2004). “Microbial Adaptation and Change: Avian Influenza,” Revue Scientifique et technique, 23(2) 453-65.
Publisher – Google Scholar